Joule-Heating Synthesis of High-Entropy Oxide Nanoparticles as Sulfion Oxidation Catalysts for Efficient and Durable Hybrid Seawater Electrolysis
焦耳热合成高熵氧化物纳米粒子作为硫离子氧化催化剂用于高效耐用的混合海水电解
第一作者: Weifeng Su, Tao-Jing Huang, Haoliang Huang
通讯作者: Guang-Jie Xia*, Zuxin Chen*, Lifeng Liu*
华南师范大学,松山湖材料实验室,大湾区大学
DOI: 待补充 | 期刊名称: 待补充 | 发表年份: 2025
PDF原文
论文亮点
- 开发了一种超快焦耳热方法合成碳负载的高熵氧化物纳米粒子(FeCoNiMnCuO HEO NPs),作为高效的硫离子氧化反应(SOR)催化剂
- 该催化剂在天然海水中表现出卓越的SOR性能,在0.545 V vs. RHE下达到500 mA cm⁻²的电流密度,并能稳定运行超过100小时
研究背景
- 直接海水电解制氢面临高能耗和竞争性氯析出反应(CER)的挑战,CER会降低效率并产生腐蚀性副产物
- 用硫离子氧化反应(SOR)替代析氧反应(OER)可规避CER问题,但实际应用需要能在高电流密度下工作的高效粉末状、负载型SOR催化剂
- 目前大多数SOR催化剂直接生长在镍泡沫或其他自支撑基底上,难以规模化生产,且在高电流密度下易形成脆性金属硫化物导致阳极失效
研究方法
- 采用超快焦耳加热法合成FeCoNiMnCuO高熵氧化物纳米粒子:将前体金属盐(Fe、Co、Ni、Mn、Cu的硝酸盐)等摩尔比负载在酸处理过的炭黑上,冻干后将粉末包裹在柔性碳纸中,通过焦耳加热系统在约1000°C下超快还原(50毫秒),随后在环境条件下自然氧化
- 使用X射线衍射(XRD)、透射电子显微镜(TEM)、高角度环形暗场扫描透射电子显微镜(HAADF-STEM)对催化剂形貌和结构进行表征
- 通过同步辐射X射线吸收光谱(XAS)和原位拉曼光谱研究催化剂的电子结构和反应机理
- 使用三电极系统在H型电解池中评估SOR性能,电解液为含2.0 M Na₂S的预处理天然海水
- 采用密度泛函理论(DFT)计算研究高熵效应对SOR性能的影响
- 构建膜电极组装体(MEA)并在单电池电解槽中测试整体海水电解性能
主要结论
超快焦耳热法成功合成了碳负载的FeCoNiMnCuO高熵氧化物纳米粒子,在碱性天然海水中表现出卓越的SOR性能
原位XAS和拉曼光谱研究表明,真正的催化活性物种是FeCoNiMnCuO NPs在S²⁻阴离子存在下表面形成的金属硫化物层,特别是Fe-S和Cu-S物种
DFT计算表明高熵效应通过增加本征电荷稳定性降低了SOR势决步骤的自由能成本,使FeCoNiMnCuO成为最活跃的SOR催化剂
催化剂形貌与结构表征
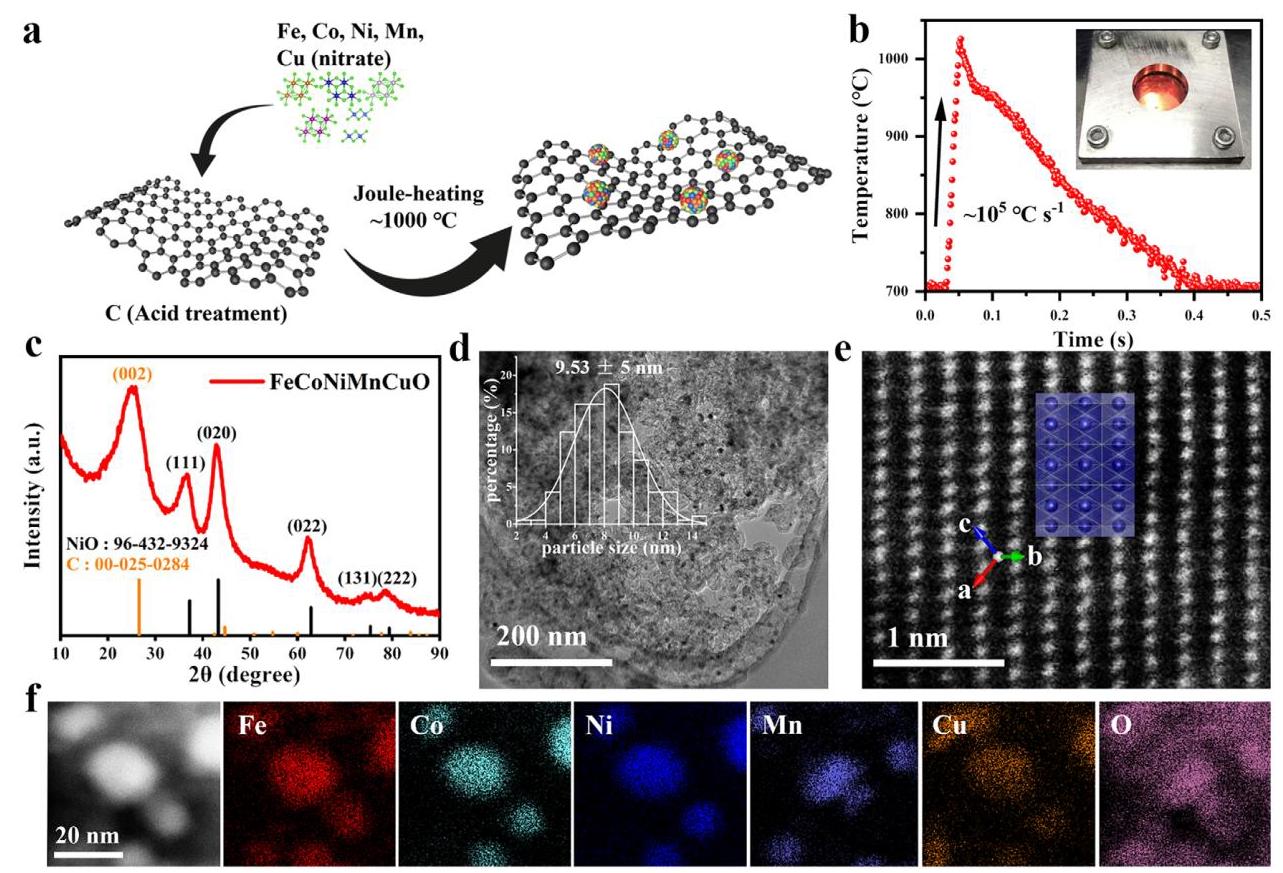
图1: a) FeCoNiMnCuO HEO NP催化剂的合成示意图; b) 焦耳加热过程中温度随时间变化的曲线; c) FeCoNiMnCuO HEO催化剂的XRD图谱; d) FeCoNiMnCuO HEO催化剂的TEM图像,插图为尺寸分布直方图; e) 沿(121)晶面投影的Cs校正STEM图像; f) HAADF-STEM图像及相应的元素分布图
分析结果: 通过超快焦耳加热法成功合成了平均尺寸为9.53±5 nm的FeCoNiMnCuO高熵氧化物纳米粒子,均匀分布在碳载体上。XRD分析表明形成了类似NiO岩盐结构的单相高熵氧化物。元素分布图显示所有元素在单个纳米粒子中基本均匀分布,证实了高熵特性的形成。
催化剂电子结构分析
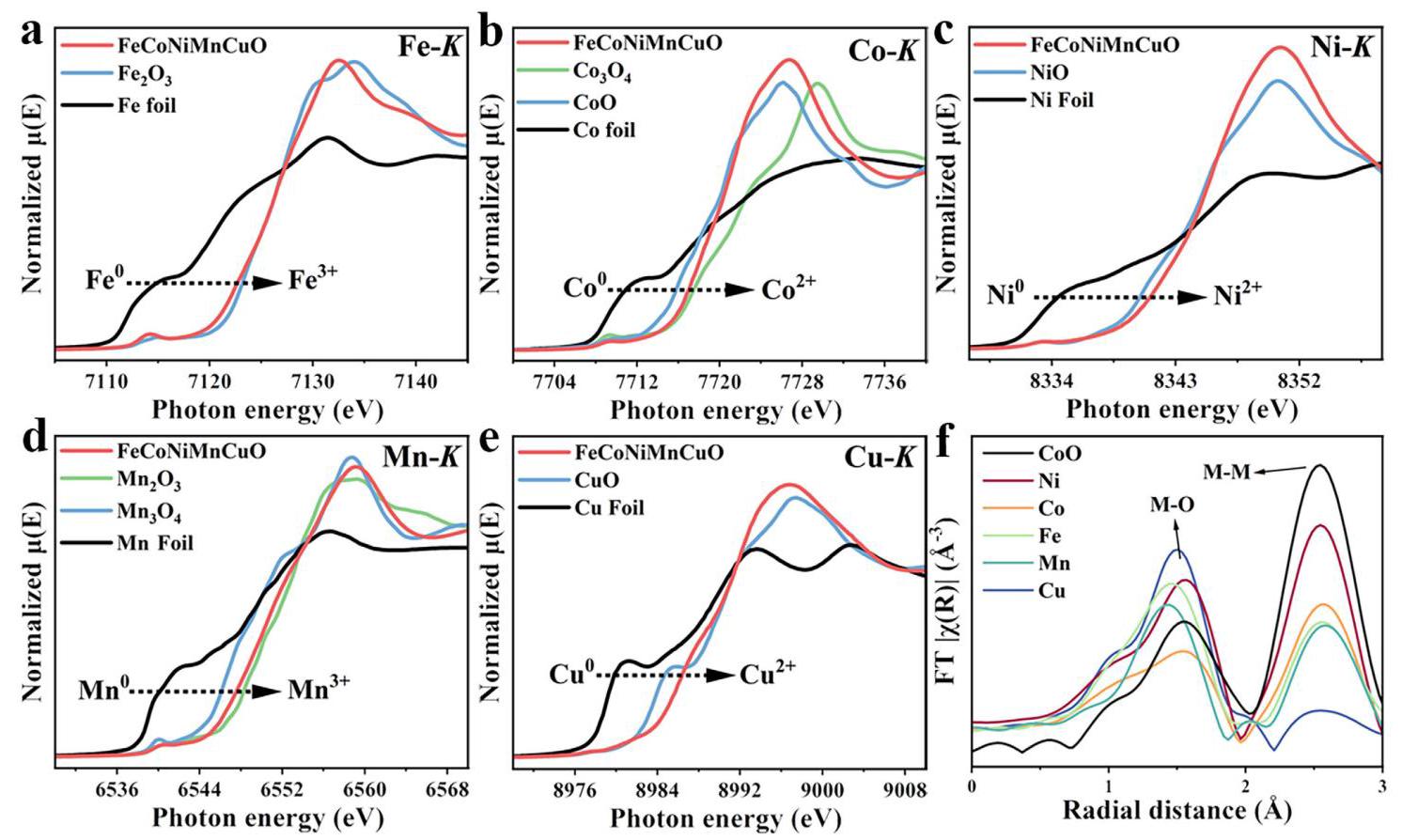
图2: a-e) FeCoNiMnCuO HEO NP催化剂在相应金属K边的XANES谱图; f) FeCoNiMnCuO HEO NP催化剂在相应金属K边的k²加权FT-EXAFS谱图
分析结果: X射线吸收近边结构(XANES)分析表明FeCoNiMnCuO中Fe、Co、Ni、Mn、Cu的氧化态分别为+3、+2、+2、+3和+2。扩展X射线吸收精细结构(EXAFS)分析显示所有五种金属具有相似的M-M散射路径长度(约3.0 Å),表明它们良好混合并采用类似CoO和NiO的岩盐结构,与XRD结果一致。
SOR电催化性能
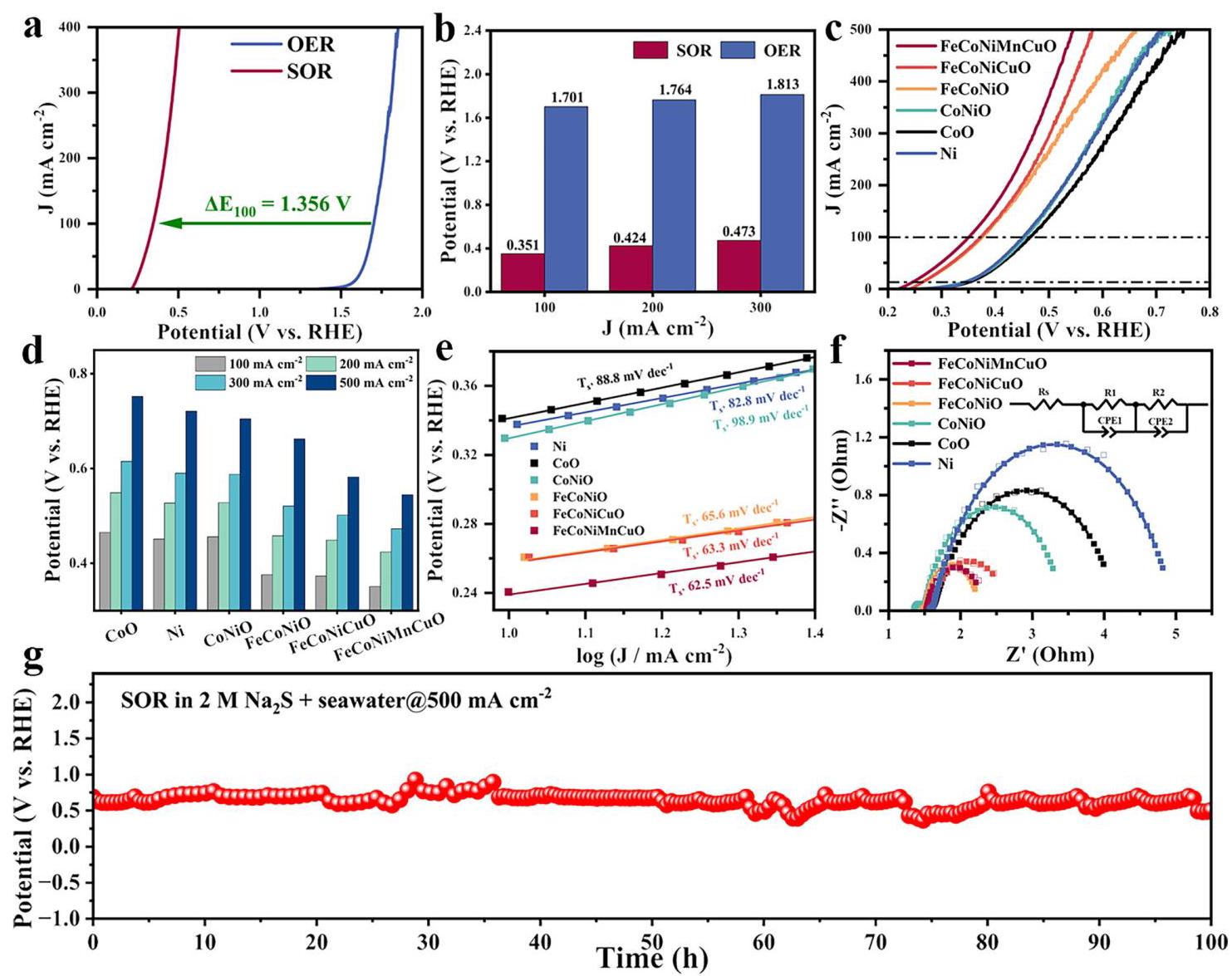
图3: a) FeCoNiMnCuO HEO NPs在碱性海水中的SOR和OER的LSV曲线; b) FeCoNiMnCuO HEO NP催化剂在SOR和OER过程中达到100、200和300 mA cm⁻²所需过电位的比较; c) 不同催化剂在2.0 M Na₂S + 海水中的SOR极化曲线; d) FeCoNiMnCuO HEO NP催化剂在不同电流密度下SOR所需电位的比较; e) 不同催化剂在SOR过程中的Tafel图; f) FeCoNiMnCuO HEO NP催化剂在0.44 V vs. RHE下测得的SOR奈奎斯特图; g) FeCoNiMnCuO HEO NP催化剂在500 mA cm⁻²下的长期催化稳定性评估
分析结果: FeCoNiMnCuO HEO NPs表现出最佳的SOR活性,仅需0.545 V vs. RHE即可达到500 mA cm⁻²的高电流密度,优于其他中熵和单/双金属氧化物催化剂。Tafel斜率(62.5 mV dec⁻¹)和电荷转移电阻均最低,表明电荷转移动力学更快。在500 mA cm⁻²下能稳定催化SOR约100小时而无明显降解,表现出卓越的长期稳定性。
原位表征与反应机理研究
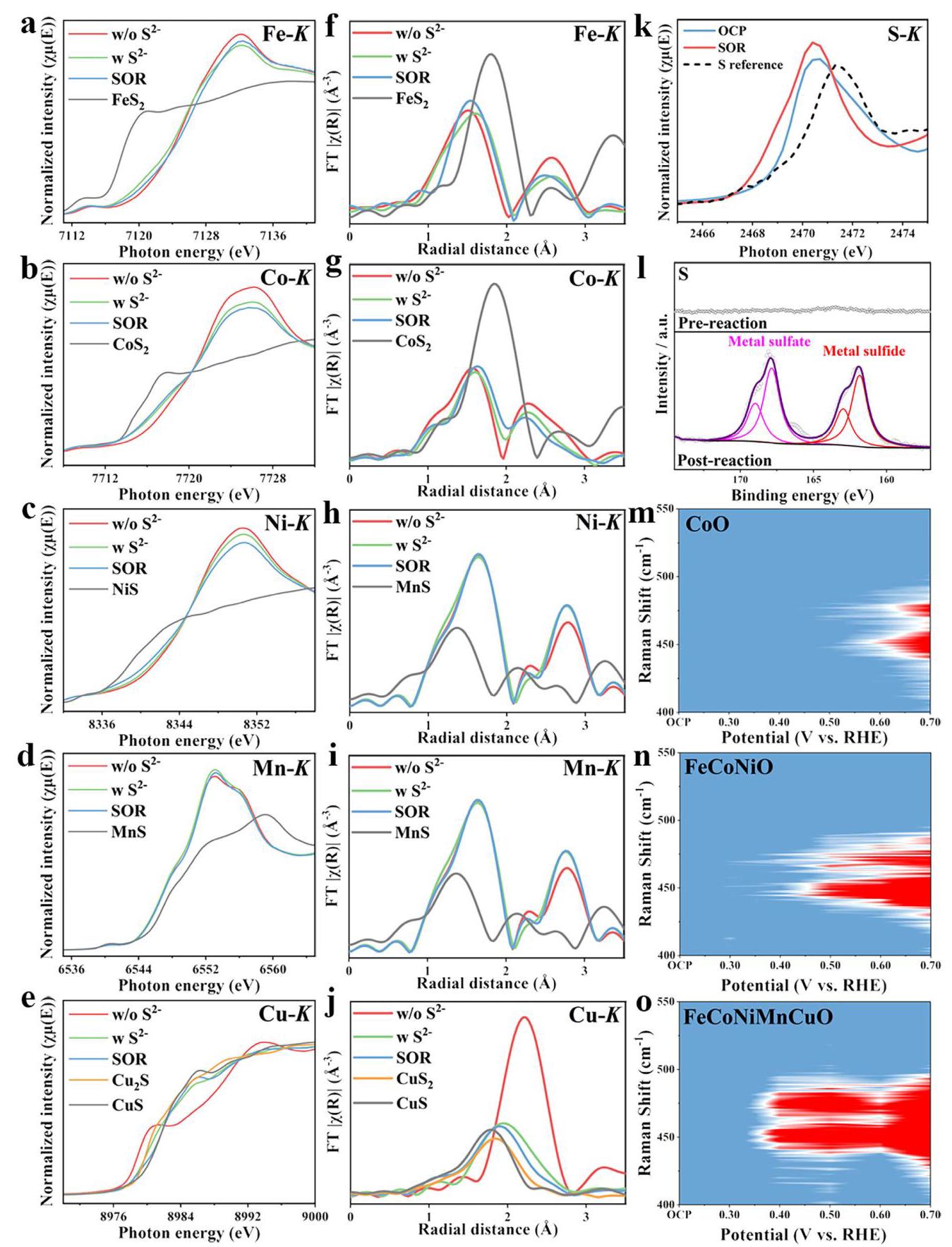
图4: a-e) FeCoNiMnCuO HEO NPs中不同过渡金属的操作金属K边XANES谱图; f-j) 相应的操作k²加权FT-EXAFS谱图; k) 在OCP和SOR后获得的催化剂S K边准原位XANES谱图; l) SOR前后的高分辨率S 1s XPS谱图; m-o) 在CoO、FeCoNiO和FeCoNiMnCuO NP催化剂上收集的2D原位拉曼光谱图
分析结果: 操作XAS研究表明,当FeCoNiMnCuO NPs与S²⁻阴离子接触时,会自发形成金属硫化物,其中Cu主要转化为Cu₂S(在OCP)和CuS(在SOR),Fe、Co和Ni也有部分硫化物生成,而Mn保持不变。真正的SOR催化活性物种是这些原位形成的金属硫化物,特别是Fe-S和Cu-S物种。原位拉曼光谱显示FeCoNiMnCuO HEO在0.4 V vs. RHE的较低电位下就出现 polysulfide 特征峰,证实了其较低的过电位和更快的反应动力学。
DFT理论计算
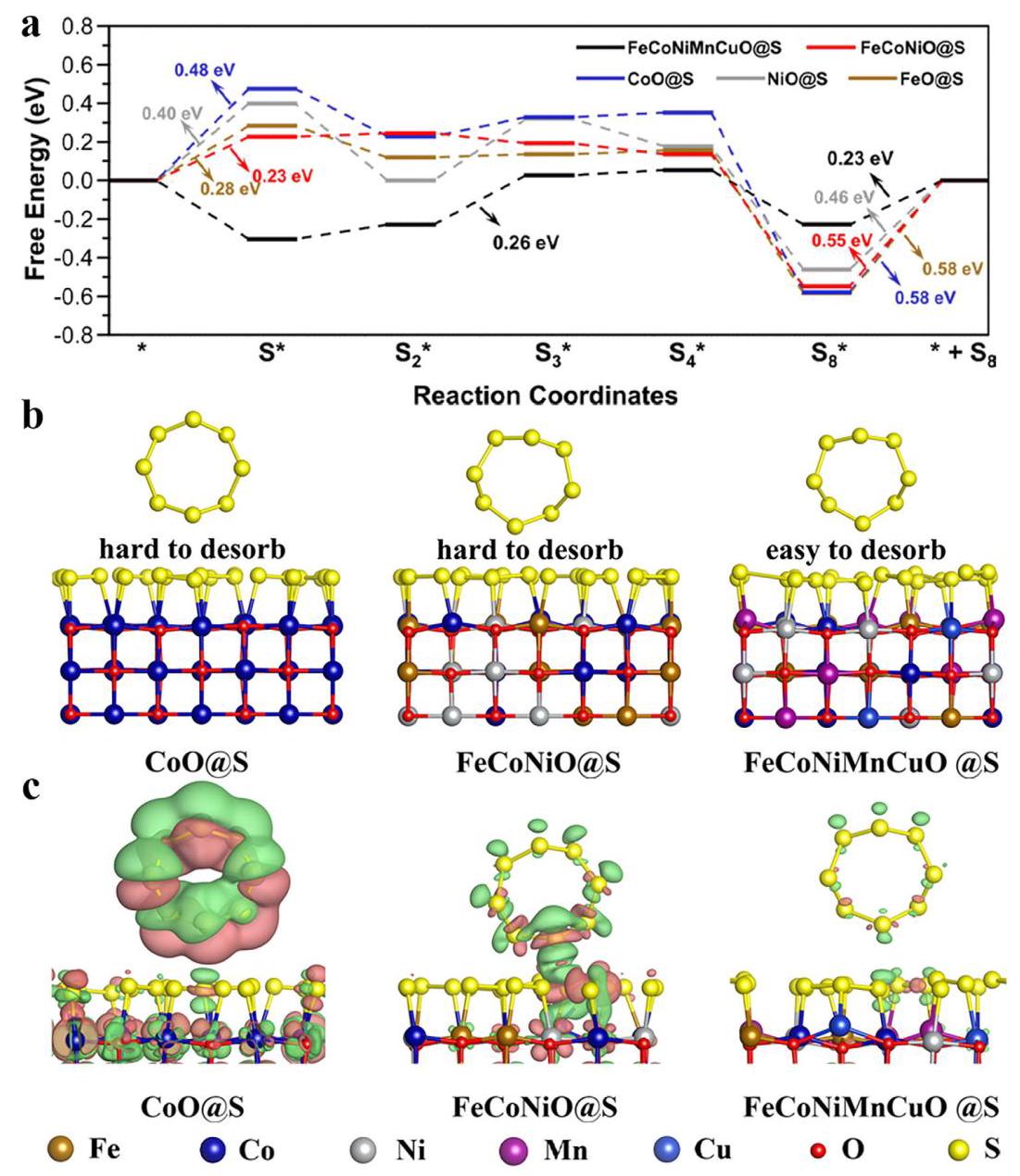
图5: a) 在不同电催化剂上SOR过程的自由能分布图; b) S₈在不同催化剂上解吸的3D构型; c) 显示S₈解吸过程中S₈与CoO@S、FeCoNiO@S和FeCoNiMnCuO@S催化剂之间电子交换的电荷密度图
分析结果: DFT计算表明,所有催化剂的势决步骤(PDS)都是S₈产物的艰难解吸。FeCoNiMnCuO@S的解吸自由能显著降低(0.23 eV),远低于其他催化剂(0.46-0.58 eV)。电荷分布分析显示,在S₈解吸过程中,FeCoNiMnCuO@S表现出更高的电荷稳定性,表面硫层的Bader电荷变化绝对值最小(0.08 |e|)。这表明高熵效应降低了SOR势决步骤的自由能成本,同时增加了内部电荷稳定性,使FeCoNiMnCuO成为最活跃的SOR催化剂。
整体海水电解性能
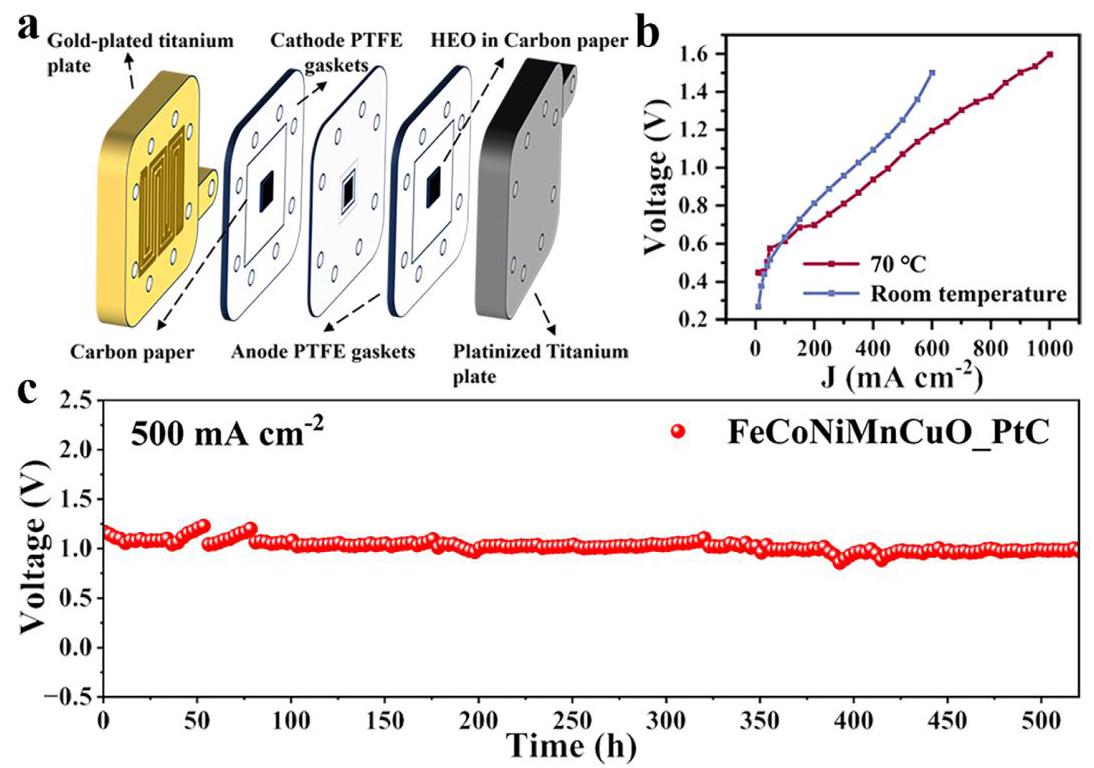
图6: a) 不对称海水电解槽示意图; b) 在室温和70°C下记录的极化曲线; c) 电解槽在70°C、500 mA cm⁻²下的长期运行稳定性评估
分析结果: 使用FeCoNiMnCuO HEO NPs作为阳极催化剂和商用Pt/C作为阴极催化剂构建的MEA电解槽,在70°C下仅需1.07 V即可提供500 mA cm⁻²的电流密度,相当于每立方米H₂仅消耗2.4 kWh的能量,显著低于传统海水电解(>4.5-6.0 kWh m⁻³ H₂)。更重要的是,该SOR辅助海水电解系统在70°C下表现出优异的长期稳定性,能在500 mA cm⁻²下连续运行超过500小时而无性能衰减,在操作电流密度和耐久性方面代表了显著进步。