Upcycling Spent Cathodes from Li-Ion Batteries into a High-Entropy Alloy Catalyst with Reverse Electron Transfer for Li-O2 Batteries
将废旧锂离子电池正极升级回收为具有反向电子转移的高熵合金催化剂用于Li-O2电池
第一作者: Peng Wang, Shan Guo, Yongbin Xu, Xinyi Yuan, Yu Tian, Binchao Xu, Zhijun Zhao, Yuxiao Wang, Jianwei Li, Xiaojun Wang
通讯作者: Peng Wang*, Zhiming Liu*
所属大学: 青岛科技大学机电工程学院,山东省高性能碳材料制备与应用工程实验室
DOI: https://doi.org/10.1021/acsnano.5c00704
PDF原文
期刊名称: ACS Nano
发表年份: 2025
论文亮点
- 开发了一种通过快速焦耳加热一步合成超细高熵合金(HEA)纳米颗粒的通用方法,该合金锚定在氮掺杂碳(N-C)载体上,作为Li-O2电池的高效催化剂。
- 实验和计算结果表明,在快速非平衡热冲击驱动下,发生了非常规的电子反向转移(从电负性更高的Pt转移到周围的NiCoMn原子),优化了Pt的电子结构,赋予了其优异的双功能催化活性。
研究背景
- 锂氧(Li-O2)电池具有极高的理论能量密度(3500 Wh kg-1),是下一代高能量密度储能设备的有力竞争者。然而,其实际应用受限于放电产物Li2O2的绝缘性和难溶性导致的氧还原反应(ORR)和氧析出反应(OER)动力学缓慢等问题。
- Pt族贵金属催化剂因其优异的电荷转移速率和可调的电子构型被认为是先进的Li-O2电池电催化剂,但其稀缺性和循环不稳定性限制了其发展。与经济的3d过渡金属(M)形成固溶体合金可以降低成本并实现电子结构调控。
- 高熵合金(HEAs)由4-5种或更多金属元素以相近摩尔比组成,因其多元素协同、晶格应变和鸡尾酒效应等产生的特殊配位和电子结构而备受关注。废旧锂离子电池正极中含有大量未填满价层d轨道的Ni、Co、Mn等有价金属,是构建PtM HEA的理想过渡金属源。
研究方法
- 材料制备:
- N-C载体制备: 将柠檬酸钠和尿素前体按10:1比例充分混合研磨,然后在氩气气氛下于750°C管式炉中煅烧1小时,经过滤、洗涤、干燥后得到多孔N-C基底。
- NCM提取物制备: 将来自电动汽车的废LiNi1/3Mn1/3Co1/3O2电池组放电至安全电压后手动拆解。将回收的正极材料在N-甲基-2-吡咯烷酮(NMP)中超声溶解PVDF粘结剂,分离出正极材料粉末。用草酸溶液处理提取锂离子,再用硝酸溶液溶解,离心去除炭黑等不溶杂质,得到含有回收的Ni、Co、Mn离子的硝酸液体上清液(NCM提取物)。
- 催化剂合成 (以Pt HEA@N-C为例): 将N-C基底、NCM提取物和H2PtCl6·6H2O按Pt:Ni:Co:Mn原子比1:1:1:1分散在无水乙醇中,室温搅拌12小时。收集沉淀并干燥后,通过焦耳加热装置快速升温至900°C,随后急速冷却至室温,诱导多元素混合,得到Pt HEA@N-C复合材料。作为对比,采用相同步骤(不添加NCM提取物或H2PtCl6·6H2O)制备了Pt@N-C和NCM@N-C催化剂。同样方法合成了Ir HEA@N-C和Ru HEA@N-C。
- 材料表征: 使用扫描电子显微镜(SEM)、透射电子显微镜(TEM)、高角度环形暗场扫描透射电子显微镜(HAADF-STEM)、X射线衍射(XRD)、X射线光电子能谱(XPS)、氮气吸附脱附测试(BET)、拉曼光谱(Raman)、X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)等技术对材料的形貌、结构、元素分布和电子结构进行了详细表征。
- 电化学测试: 将催化剂、导电炭黑(Super C65)和聚偏氟乙烯(PVDF)按80:10:10的质量比在NMP中混合制成浆料,涂覆在碳纸上,干燥后作为工作阴极。组装2032型纽扣电池(锂阳极、玻璃纤维隔膜、催化剂阴极、1M LiTFSI/TEGDME电解液),在充满纯氧的密闭箱中静置后,使用电池测试系统进行恒电流充放电测试、循环性能测试。使用电化学工作站进行循环伏安(CV)和电化学阻抗谱(EIS)测试。
- 理论计算: 采用基于VASP软件的密度泛函理论(DFT)计算,分析了Pt HEA和纯Pt纳米颗粒对O2和LiO2的吸附能、吉布斯自由能路径、态密度(PDOS)、电荷密度差分以及Bader电荷等,以揭示催化机理。
主要结论
- 成功开发了一种基于废旧电池回收和快速焦耳加热的通用策略,合成了超细Pt/Ir/Ru基高熵合金纳米催化剂(Pt/Ir/Ru HEA@N-C)。
- 研究发现,在快速焦耳加热的极端热冲击驱动下,发生了从电负性更高的Pt到周围电负性较低的Ni、Co、Mn原子的非常规反向电子转移,优化了Pt活性位点的电子结构(d带中心上移)。
- 优化后的Pt HEA@N-C催化剂在Li-O2电池中表现出卓越的双功能催化活性和稳定性,实现了极低的过电位(0.27 V)和超长的循环寿命(超过240次循环)。该工作为废旧锂离子电池的升级回收和高性能电催化剂的设计提供了新思路。
催化剂制备流程与机理示意图
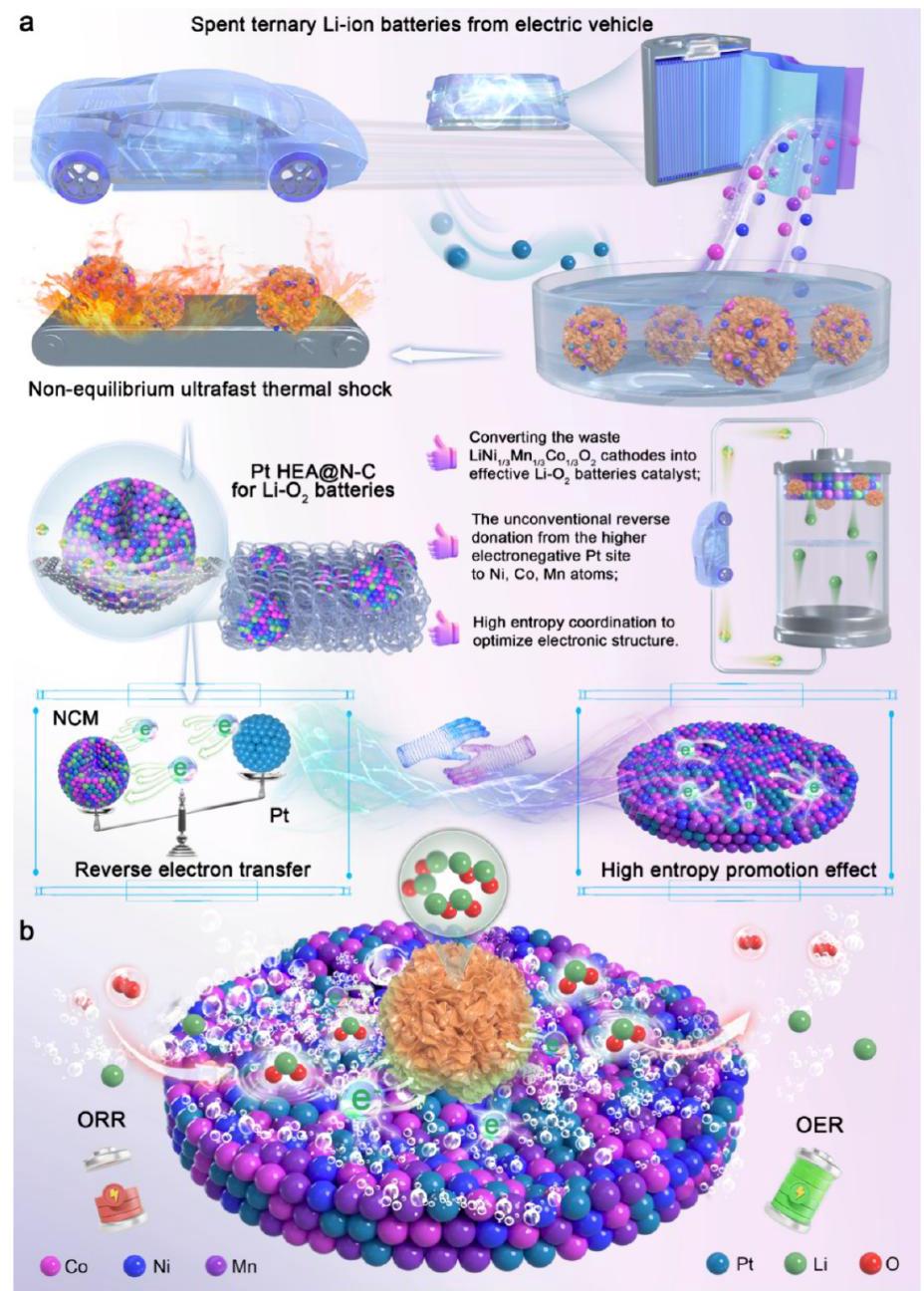
图1. (a) Pt HEA@N-C催化剂的制备流程示意图和(b) underlying mechanism示意图。
分析结果: 该图直观展示了本研究的核心策略:从废旧三元锂离子电池正极中提取Ni、Co、Mn有价金属,与Pt源前驱体混合,并通过快速的焦耳加热过程,一步合成锚定在氮掺杂碳载体上的高熵合金纳米颗粒。图b暗示了其独特的反向电子转移机理,这是其高性能的关键。
Pt HEA@N-C的形貌表征

图2. Pt HEA@N-C的形貌表征。(a) SEM图像, (b) TEM图像, (c) HAADF-STEM图像 (插图: 粒径分布), (d) HRTEM图像 (插图: Pt HEA纳米颗粒的FFT花样)。(e) (200)晶面的IFFT图像和晶粒间隙图, (f) (111)晶面的IFFT图像和晶粒间隙图。(g) (200)晶面的GPA分析, (h) (111)晶面的GPA分析。(i, j) HAADF-STEM图像 (插图: FFT花样), (k) 3D强度分布。(l) 线扫描和 (m) 相应的元素分布Mapping图像。
分析结果: 多种电子显微镜技术证实了超小的Pt HEA纳米颗粒(平均尺寸~3.36 nm)均匀分布在N-C载体上,没有团聚。HRTEM和FFT表明其具有面心立方(fcc)结构。IFFT和GPA分析揭示了由于不同金属原子半径差异和快速加热-淬火过程引起的显著晶格应变和位错缺陷。HAADF-STEM图像中的原子柱没有明显的周期性明暗对比,表明Pt原子与周围Ni、Co、Mn原子随机配位,没有成分和相分离。线扫描和元素Mapping进一步证明了Pt、Ni、Co、Mn四种元素在单个纳米颗粒内均匀分布,成功合成了单相固溶体高熵合金。
Ir/Ru HEA@N-C的形貌表征
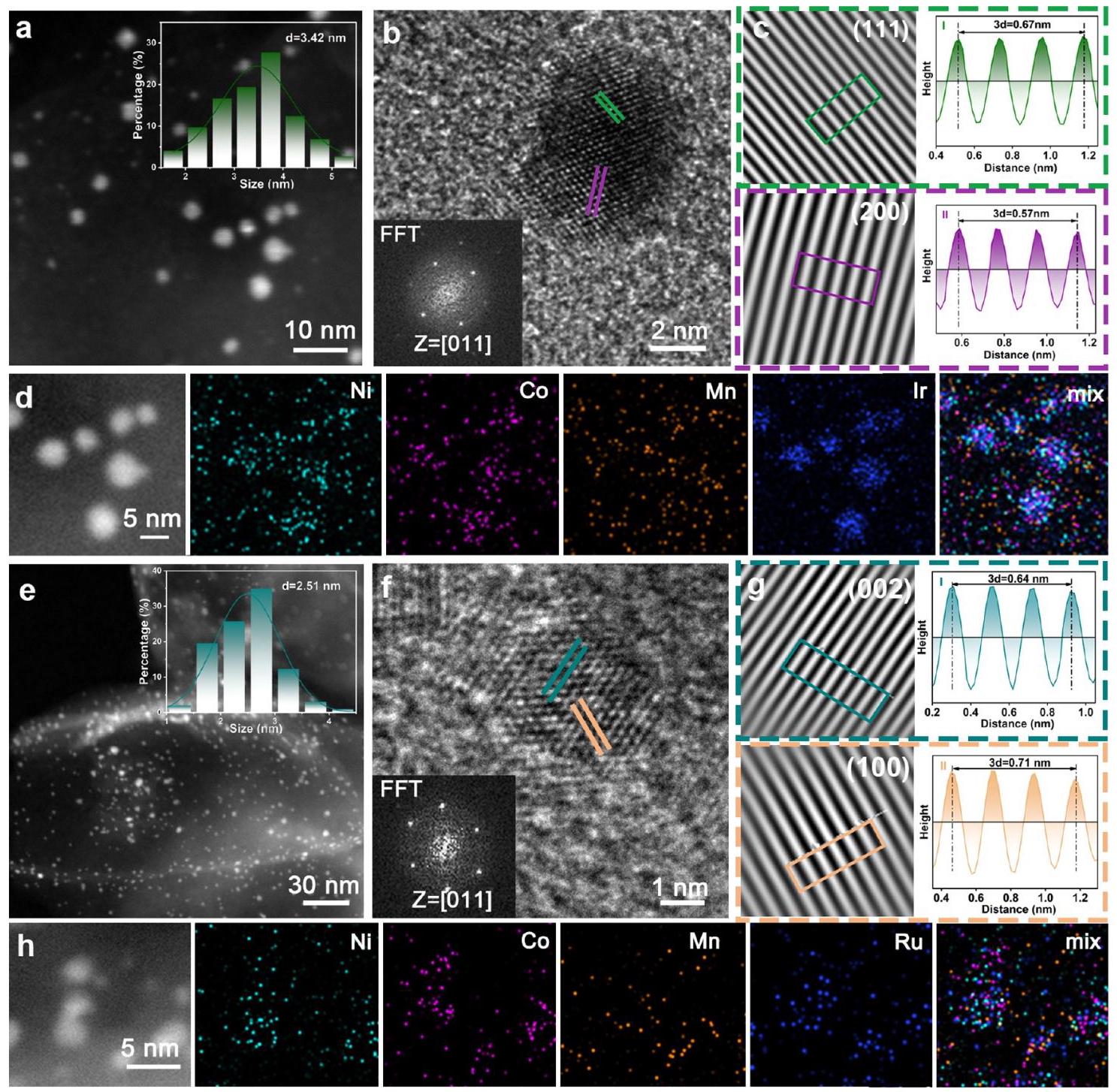
图3. Ir HEA@N-C和Ru HEA@N-C的形貌表征。(a) Ir HEA@N-C的HAADF-STEM图像 (插图: 粒径分布), (b) Ir HEA@N-C的HRTEM图像 (插图: Ir HEA纳米颗粒的FFT花样)。(c) Ir HEA纳米颗粒的(111), (200)晶面的IFFT图像和晶粒间隙图。(d) Ir HEA@N-C的元素分布Mapping图像。(e) Ru HEA@N-C的HAADF-STEM图像 (插图: 粒径分布), (f) Ru HEA@N-C的HRTEM图像 (插图: Ru HEA纳米颗粒的FFT花样)。(g) Ru HEA纳米颗粒的(002), (100)晶面的IFFT图像和晶粒间隙图。(h) Ru HEA@N-C的元素分布Mapping图像。
分析结果: 此图表明本研究提出的合成策略具有普适性,可成功扩展到其他铂族金属(Ir, Ru)高熵合金催化剂的制备。Ir HEA@N-C形成了fcc结构,平均粒径约3.42 nm。Ru HEA@N-C则形成了六方相结构,平均粒径更小(约2.51 nm)。相应的元素Mapping和线扫描(图S12)均证实了Ir/Ru、Ni、Co、Mn元素在各自合金纳米颗粒中的均匀分布,成功形成了高熵合金结构。
Pt HEA@N-C的结构与电子结构表征
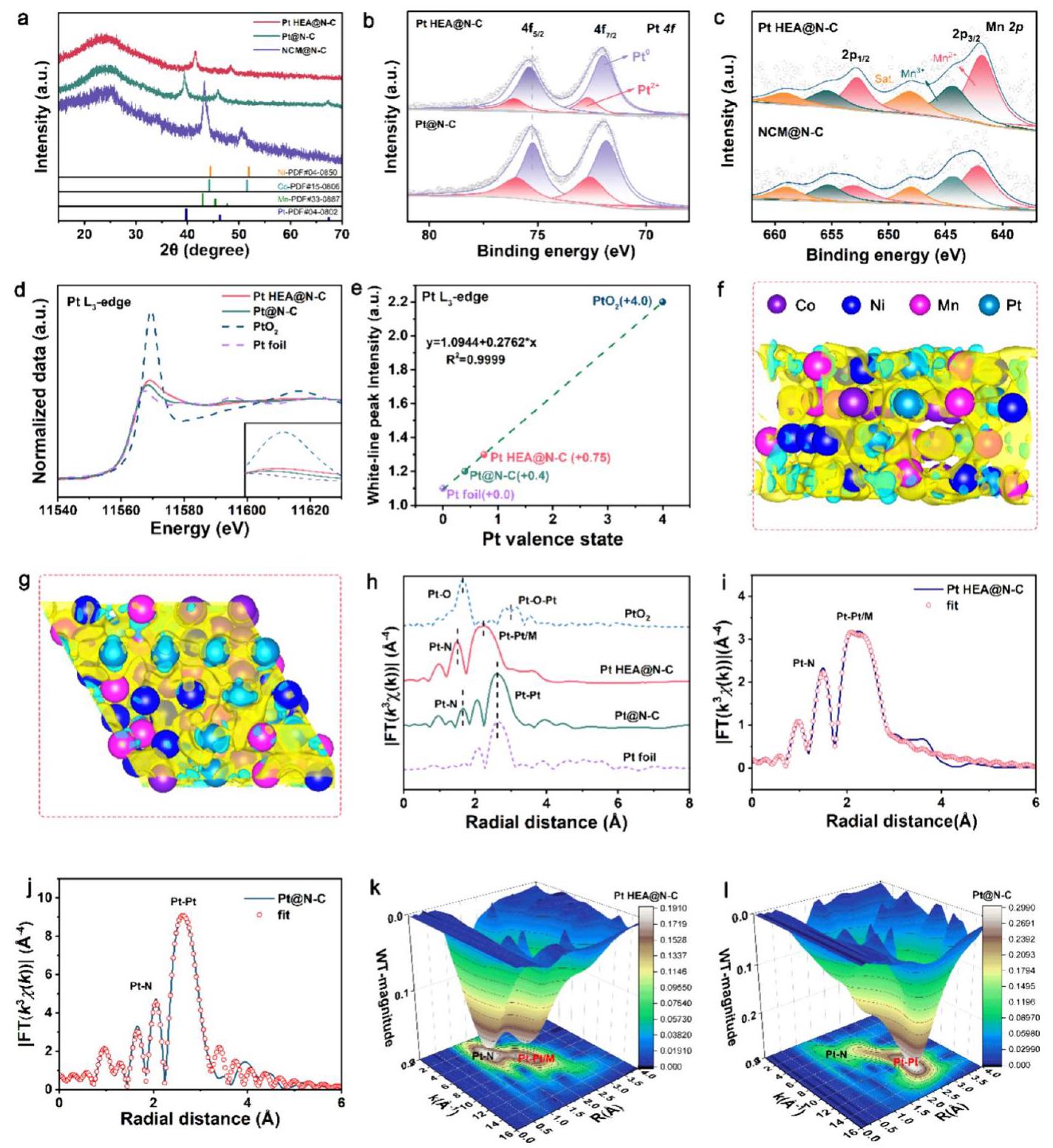
图4. Pt HEA@N-C, Pt@N-C, 和 NCM@N-C的结构表征。(a) XRD图谱。(b) Pt 4f 和 (c) Mn 2p 的高分辨率XPS光谱。(d) Pt HEA@N-C, Pt@N-C, PtO2, Pt箔的归一化L3-边XANES光谱。(e) Pt HEA和Pt@N-C中Pt的拟合价态。(f, g) Pt HEA内电荷密度差分图像的侧视图和正视图。黄色和青色轮廓分别代表电子积累和耗尽。(h) Pt HEA@N-C, Pt@N-C, PtO2, Pt箔的R空间L3-边FT-EXAFS光谱。(i) Pt HEA@N-C 和 (j) Pt@N-C的R空间EXAFS拟合曲线。(k) Pt HEA@N-C 和 (l) Pt@N-C的WT-EXAFS分析。
分析结果: 此图通过多种光谱技术深入揭示了Pt HEA@N-C的独特电子结构和配位环境。
- XPS (b, c): Pt HEA@N-C的Pt 4f结合能相对于Pt@N-C向高结合能方向移动,而Ni 2p, Co 2p, Mn 2p结合能则向低结合能方向移动,表明电子从Pt原子转移到了Ni、Co、Mn原子,发生了反向电子转移,导致Pt缺电子而Ni/Co/Mn富电子。
- XANES (d, e): Pt HEA@N-C的白线强度高于Pt@N-C,表明其Pt的氧化态更高(~+0.75),与XPS结果一致。
- DFT电荷密度差分 (f, g): 理论计算直观显示Pt位点(青色)电子耗尽,而Ni、Co、Mn位点(黄色)电子积累,支持了反向电子转移的可能性。
- EXAFS (h-j): Pt HEA@N-C的Pt-M(M=Ni,Co,Mn,Pt)键长(~2.54-2.66 Å)比Pt@N-C的Pt-Pt键长(~2.75 Å)更短,配位数也更低,证实了Pt与Ni、Co、Mn的成功合金化并形成了无序的高熵配位结构。WT-EXAFS (k,l) 进一步支持了多种元素在单相fcc HEA纳米颗粒中的均匀混溶性。
这些表征共同证实了高熵结构和反向电子转移对Pt电子结构的优化作用。
电化学性能
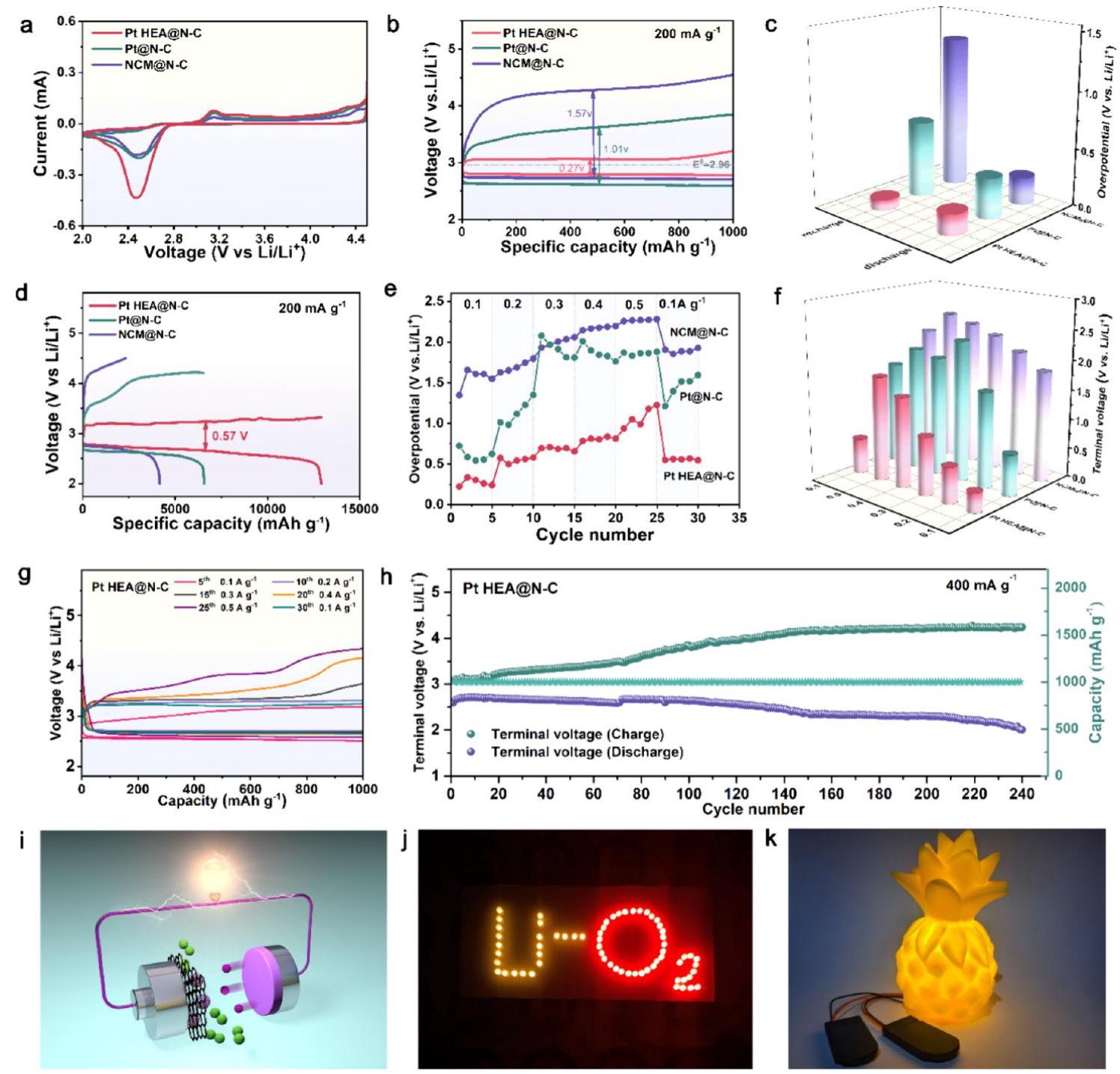
图5. Pt HEA@N-C, Pt@N-C, 和 NCM@N-C的电化学性能。(a) 扫描速率为0.1 mV s-1的CV曲线。(b) 在200 mA g-1电流密度、1000 mAh g-1截止容量下的放电-充电曲线。(c) (b)中总结的放电和充电过电位。(d) 在200 mA g-1电流密度下的首次深度放电-充电曲线。(e) 在0.1至0.5 A g-1不同电流密度、1000 mAh g-1限制容量下三种电极的过电位。(f) 三种电极在不同电流密度下的终止放电和充电电压间隙。(g) Pt HEA@N-C电极在不同电流密度下的放电-充电曲线。(h) Pt HEA@N-C电极在400 mA g-1、1000 mAh g-1下的循环稳定性。(i) Li-O2电池示意图。(j) 由配备Pt HEA@N-C催化剂的锂空气电池点亮的LED照片。(k) Pt HEA@N-C基锂空气电池成功点亮菠萝玩具。
分析结果: 该图全面展示了Pt HEA@N-C催化剂卓越的电化学性能。
- CV (a): Pt HEA@N-C电极显示出更高的还原峰电流和积分面积,以及更正的ORR起始电位和更负的OER起始电位,表明其具有更快的动力学响应和改善的O2氧化还原效率。
- 充放电曲线 (b, d): Pt HEA@N-C电极实现了极低的总过电位(0.27 V),放电和充电过电位分别仅为0.18 V和0.09 V,能量效率高达91.1%,远优于Pt@N-C(1.01 V)和NCM@N-C(1.57 V)电极。其深度放电容量高达12917.4 mAh g-1,且库伦效率为100%。
- 倍率性能 (e-g): 在不同电流密度下,Pt HEA@N-C电极均表现出最小的电压波动和电压滞后,显示出优异的高倍率催化动力学和可靠性。
- 循环性能 (h): Pt HEA@N-C电极在400 mA g-1、1000 mAh g-1条件下表现出优异的循环寿命,可稳定运行超过240次循环,远超对比样品。
- 实际应用展示 (j, k): 组装的锂空气电池成功点亮了LED阵列和菠萝玩具,证明了其实际应用的可行性。
这些结果突出了反向电子转移和高熵配位效应在优化Li
2O
2成核和分解行为、从而增强催化动力学和能量效率方面的关键作用。
放电/充电后电极的非原位表征
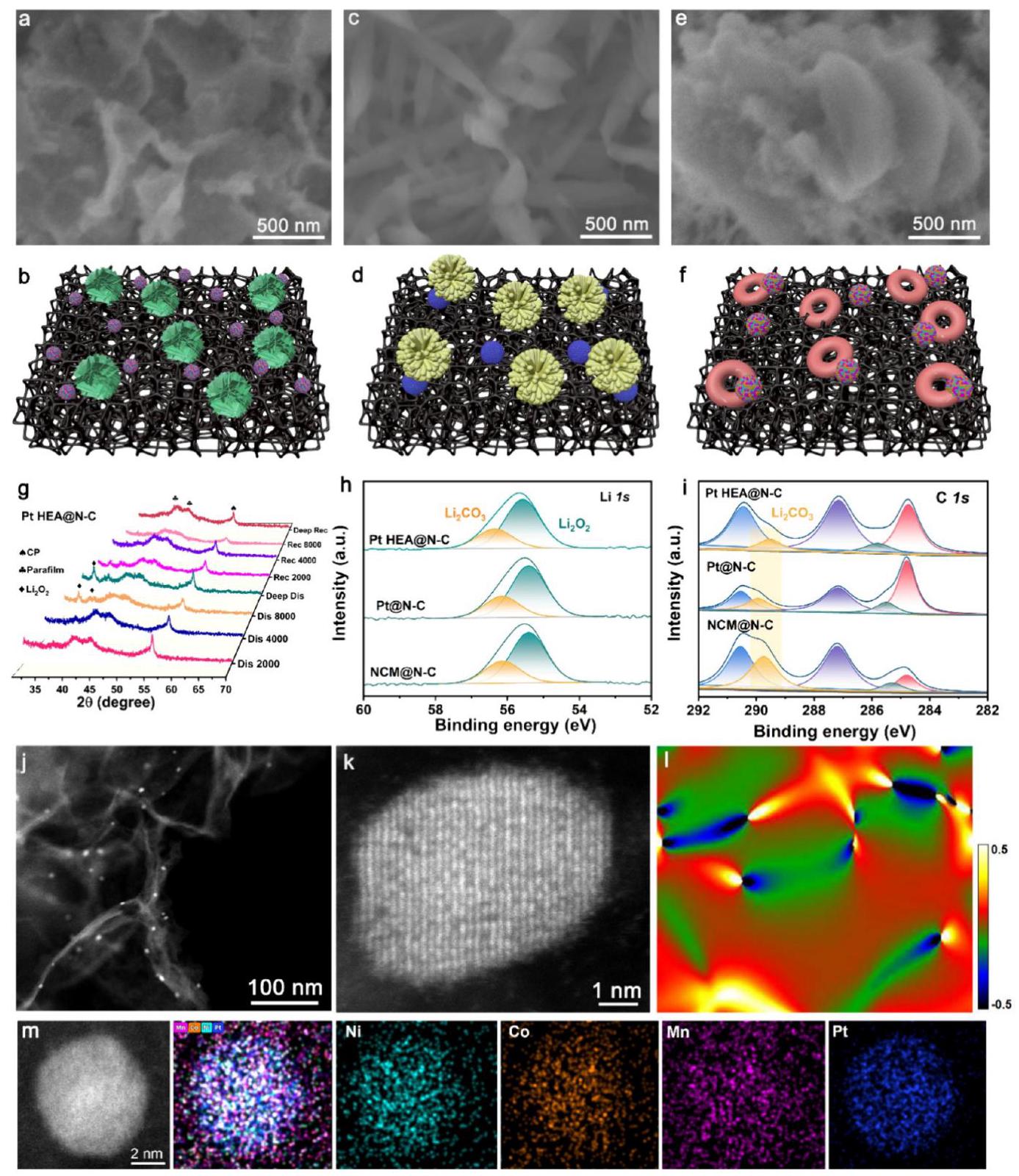
图6. 放电和充电后电极的非原位表征。(a, b) 完全放电后的Pt HEA@N-C, (c, d) Pt@N-C, 和 (e, f) NCM@N-C的非原位SEM图像和模拟示意图。(g) 第一个循环不同阶段充电和放电后Pt HEA@N-C的非原位XRD图谱。(h) 第30次循环放电后Pt HEA@N-C, Pt@N-C, NCM@N-C电极的Li 1s 和 (i) C 1s 区域的非原位XPS光谱。(j, k) 循环200次后的Pt HEA@N-C催化剂的非原位HAADF-STEM图像, (l) GPA结果, 和 (m) 元素Mapping。
分析结果: 此图通过非原位表征深入分析了催化剂的反应机理和稳定性。
- SEM (a-f): 深度放电后,Pt HEA@N-C电极表面均匀覆盖着丰富的花瓣状Li2O2,由堆叠的超薄纳米片组成,这种形貌有利于提供更大的三相界面。而Pt@N-C和NCM@N-C电极表面则分别被大量棒状和碟状Li2O2覆盖,这会削弱界面接触。充电后,Pt HEA@N-C催化剂表面能很好地恢复,没有放电产物残留,表明其高效的Li2O2分解能力。
- XRD (g): 放电后Pt HEA@N-C电极检测到Li2O2特征峰,充电后迅速减弱,表明Li2O2是主要放电产物且具有优异的转化可逆性。对比样品则存在Li2O2分解不完全的情况。
- XPS (h, i): 第30次循环放电后,Pt HEA@N-C电极表面的Li2CO3副产物含量最低(~28.2%),C 1s光谱中Li2CO3信号可忽略不计,表明其优异的副产物抑制能力。
- 循环后催化剂表征 (j-m): 即使经过200次循环,Pt HEA纳米颗粒仍然均匀分散,晶格条纹清晰可见,GPA显示仍存在晶格应变,元素分布保持均匀,无团聚或解离,证明了Pt HEA@N-C催化剂出色的长期结构和组成稳定性,这归因于单相fcc固溶体合金结构中稳健的高熵稳定效应。
DFT计算与机理分析
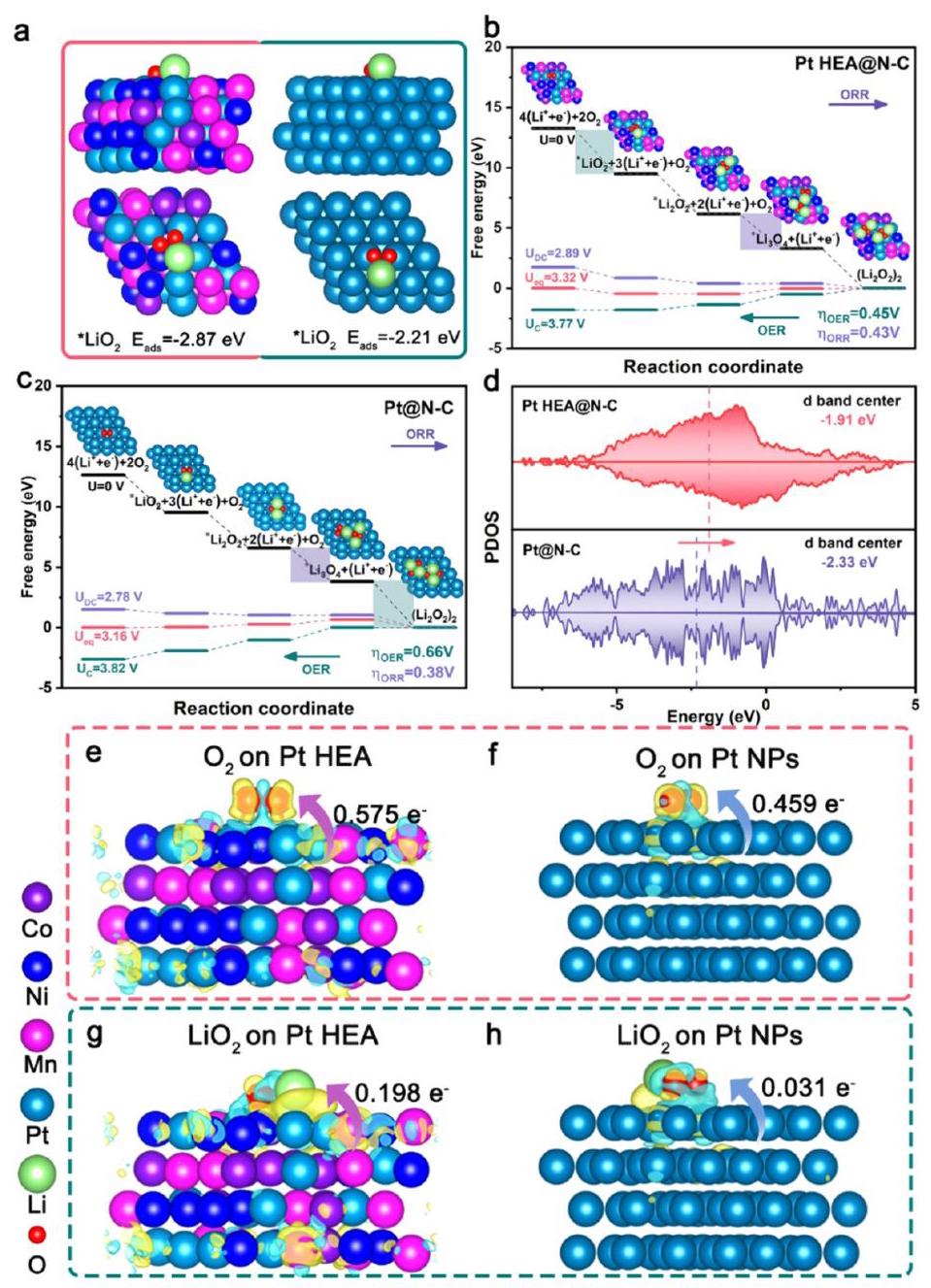
图7. DFT计算。(a) Pt HEA和Pt NPs对LiO2吸附能的比较。(b) Pt HEA 和 (c) Pt NPs的吉布斯自由能图。(d) Pt HEA和Pt NPs的PDOS图。O2在 (e) Pt HEA 和 (f) Pt NPs上吸附的电荷密度差分侧视图。LiO2在 (g) Pt HEA 和 (h) Pt NPs上吸附的电荷密度差分侧视图。黄色和青色轮廓分别代表电子积累和耗尽。
分析结果: DFT计算从理论上深入揭示了Pt HEA催化性能增强的机理。
- 吸附能 (a): Pt HEA对O2和LiO2的吸附能(-1.73 eV和-2.87 eV)均显著高于纯Pt NPs(-0.91 eV和-2.21 eV),表明Pt HEA对含氧中间体具有更强的亲和力,这有利于调控Li2O2的生长模式(形成更易分解的花瓣状而非棒状)。
- 吉布斯自由能 (b, c): 对于OER过程,Pt HEA的速率决定步骤(RDS)是LiO2氧化为O2,能垒为3.77 eV;而Pt NPs的RDS是Li4O4氧化为Li3O4,能垒为3.82 eV。计算得到的Pt HEA的OER过电位(0.45 V)低于Pt NPs(0.66 V),表明其更快的OER催化动力学。
- PDOS (d): Pt HEA的d带中心(-1.91 eV)相较于Pt NPs(-2.33 eV)上移了0.42 eV,更接近费米能级。d带中心的上移通常意味着对反应中间体的吸附能力增强,与吸附能结果一致。
- 电荷密度差分 (e-h): 直观显示了催化剂基底与吸附的含氧物种之间通过金属-氧键建立了有效的电荷转移路径。计算得到的Pt HEA表面吸附O2和LiO2的电荷转移数(0.575和0.198)远大于Pt NPs(0.459和0.031),表明Pt HEA对中间体具有更突出的亲和力和加速的电子转移趋势。
理论计算结果表明,反向电子转移和高熵配位环境优化了Pt的电子结构,增强了对关键中间体的吸附,降低了反应能垒,从而显著提升了ORR和OER动力学。