MoZn-based high entropy alloy catalysts enabled dual activation and stabilization in alkaline oxygen evolution
基于MoZn的高熵合金催化剂在碱性析氧反应中实现双重激活和稳定
Yunjie Mei1†, Jinli Chen1†, Qi Wang2†, Yaqing Guo1,3, Hanwen Liu1, Wenhui Shi1, Cheng Lin1, Yifei Yuan3, Yuhua Wang4*, Bao Yu Xia5*, Yonggang Yao1*
DOI: 10.1126/sciadv.adq6758 | Science Advances | 2024
PDF原文
论文亮点
- 开发了MoZnFeCoNi高熵合金催化剂,成功实现了吸附物演化机制(AEM)和晶格氧机制(LOM)的双重激活,突破了传统催化剂在活性和稳定性之间的权衡
- 该催化剂表现出卓越的电催化性能:在10 mA cm⁻²电流密度下过电位仅为221 mV,在100 mA cm⁻²高电流密度下稳定运行超过1500小时,远超商业IrO₂催化剂和其他高熵合金阳极
研究背景
- 电化学水分解需要高活性、长寿命且成本低廉的催化剂,以满足大规模绿色制氢和可再生能源存储的需求,特别是对于缓慢且腐蚀性的析氧反应(OER)
- 目前OER存在两种主要反应机制:吸附物演化机制(AEM)受线性标度关系限制导致动力学缓慢,而晶格氧机制(LOM)则因晶格氧逃逸导致结构不稳定
- 高熵合金(HEA)催化剂因其独特的高熵效应、晶格畸变、缓慢扩散和"鸡尾酒效应"而成为新兴材料平台,但含有易挥发和易反应元素(如Mo和Zn)的HEA制备仍面临重大挑战
研究方法
时空限制合成策略
采用金属有机框架(MOF)前驱体实现空间限制,结合快速热冲击热解(1200°C, 0.1秒)实现时间限制,成功合成MoZn基高熵合金:
- 使用2,5-二羟基对苯二甲酸(H₄DOBDC)配体与Ni²⁺、Fe³⁺、Co²⁺、MoO₄⁻和Zn²⁺组装形成高熵MOF前驱体
- 通过碳热冲击(CTS)热解实现瞬间MOF分解和碳化,短时间处理避免金属蒸发和碳化物形成
- 制备对照样品:仅时间限制(T-HEA)和仅空间限制(S-HEA)
材料表征技术
- X射线衍射(XRD)分析晶体结构
- X射线光电子能谱(XPS)和电感耦合等离子体发射光谱(ICP-OES)分析元素组成
- N₂吸附-脱附等温线测定BET比表面积
- 扫描电子显微镜(SEM)、透射电子显微镜(TEM)和能量色散X射线光谱(EDS)观察形貌和元素分布
电化学性能测试
- 三电极系统在N₂饱和的1.0 M KOH溶液中测试OER性能
- 线性扫描伏安法(LSV)测定极化曲线
- 电化学阻抗谱(EIS)分析电荷转移现象
- 计时电位法(CP)评估长期稳定性
理论计算
- 密度泛函理论(DFT)计算研究OER机理
- 分析AEM和LOM路径的能量壁垒
- 计算元素间形成能评估合金稳定性
主要结论
- 成功开发了时空限制合成策略,制备出具有分级多孔结构和3D碳框架的MoZn基高熵合金催化剂,实现了OER活性和稳定性的同步提升
- DFT计算和化学探针实验证实了MoZn基HEA催化剂中AEM和LOM的双重激活:Co-Co*-Mo位点促进AEM实现快速去质子化,而Zn-O*-Ni位点触发LOM实现高效O-O耦合
- 多元素相互作用、熵稳定和3D碳网络对整体结构的稳定至关重要,使催化剂在100 mA cm⁻²高电流密度下稳定运行超过1500小时,显著优于现有报道的大多数催化剂
图1: MoZn基HEA在OER中的双重激活和稳定设计
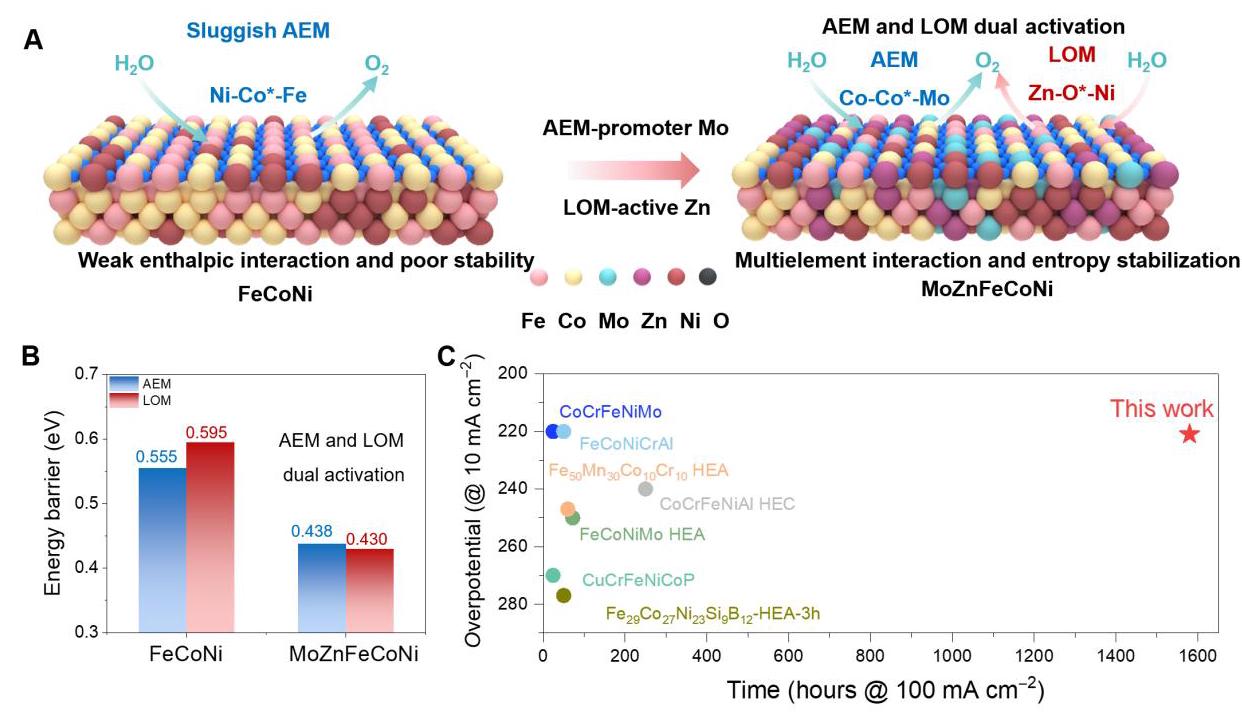
图1. MoZn基HEA在OER中的双重激活和稳定设计。(A) FeCoNi显示缓慢AEM和具有AEM和LOM双重激活的MoZn基HEA示意图。(B) 不同OER机制中计算的极限能垒,证明MoZnFeCoNi中的双重激活。(C) 根据近期文献对催化剂活性和稳定性的比较分析。
分析结果
图1A展示了FeCoNi和MoZn基HEA的机理示意图,直观比较了传统催化剂的单一机制与新型催化剂的双重机制优势。图1B的DFT计算结果表明,MoZnFeCoNi对AEM和LOM的能量壁垒(分别为0.438 eV和0.430 eV)均显著低于FeCoNi(0.555 eV和0.595 eV),证实了双重激活效应。图1C的对比分析显示,MoZn基HEA在活性和稳定性方面均优于其他高熵合金阳极和商业催化剂,展示了其卓越的综合性能。
图2: MoZn基HEA及对照样品的合成与结构表征
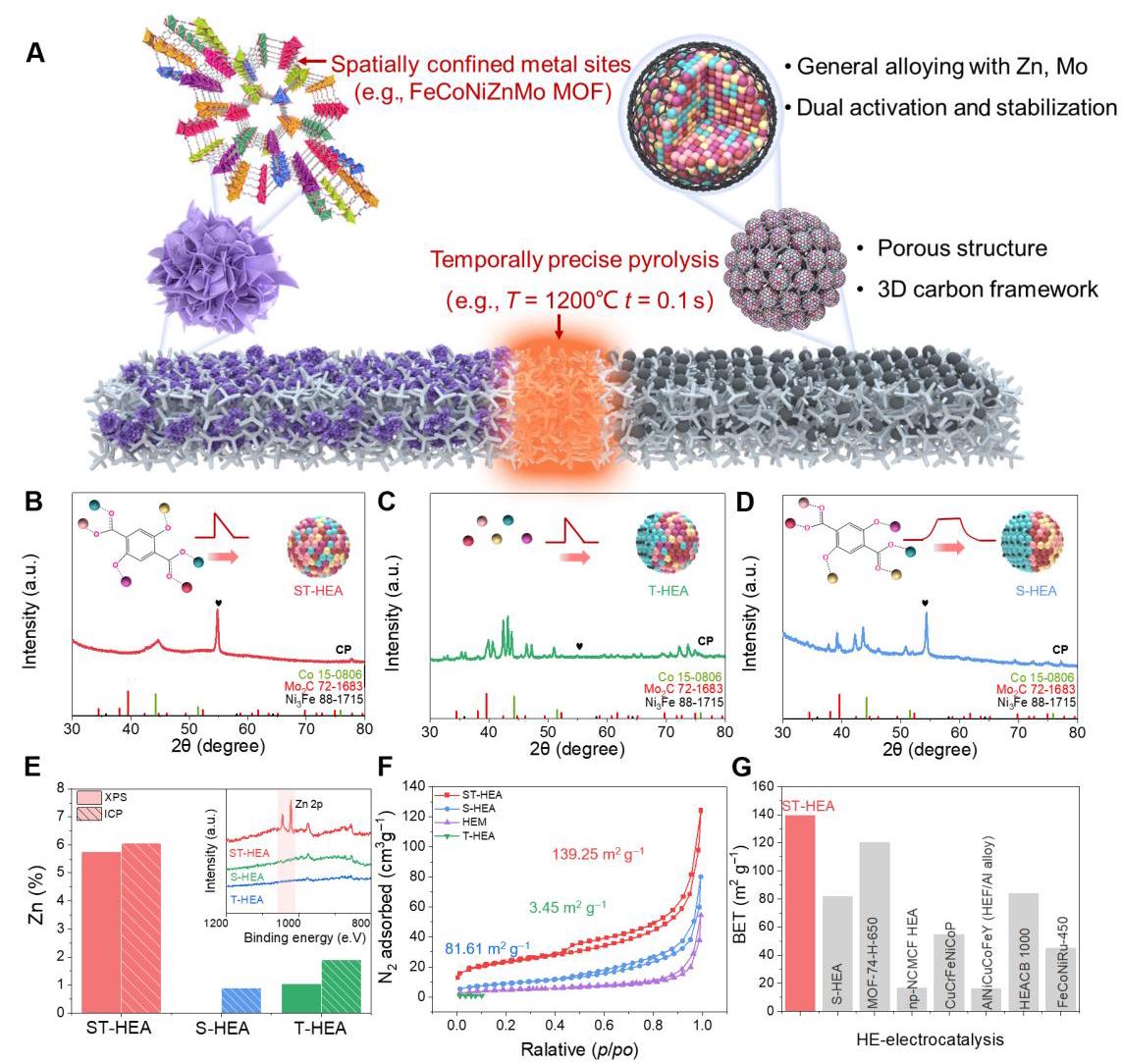
图2. MoZn基HEA及对照样品的合成与结构表征。(A) 考虑到易反应的Mo和Zn,时空限制合成在MoZn基HEA催化剂合成中具有优势。(B-D) ST-HEA、T-HEA和S-HEA的XRD分析。(E) 通过X射线光电子能谱(XPS)和电感耦合等离子体发射光谱(ICP-OES)测量的Zn含量。(F) 不同样品的N₂吸附-脱附等温线和(G) 近期文献报道的高熵催化剂的BET值。
分析结果
图2A展示了时空限制合成策略的示意图,该策略成功解决了Mo和Zn在高温合成过程中的挥发和反应问题。XRD分析(图2B-D)显示,只有ST-HEA呈现出清晰的面心立方(FCC)结构,而没有适当空间或时间限制的T-HEA和S-HEA则出现明显的第二相。图2E的XPS和ICP-OES结果表明,时空限制合成能最大程度地保留挥发性Zn元素(ST-HEA: 5.75%, T-HEA: 1.05%, S-HEA: 0%)。图2F-G的BET分析显示,ST-HEA具有极高的比表面积(139.25 m²/g),远高于T-HEA(3.45 m²/g)和S-HEA(81.61 m²/g),这得益于MOF快速热解继承的3D多孔结构。
图3: MoZn基HEA的详细表征
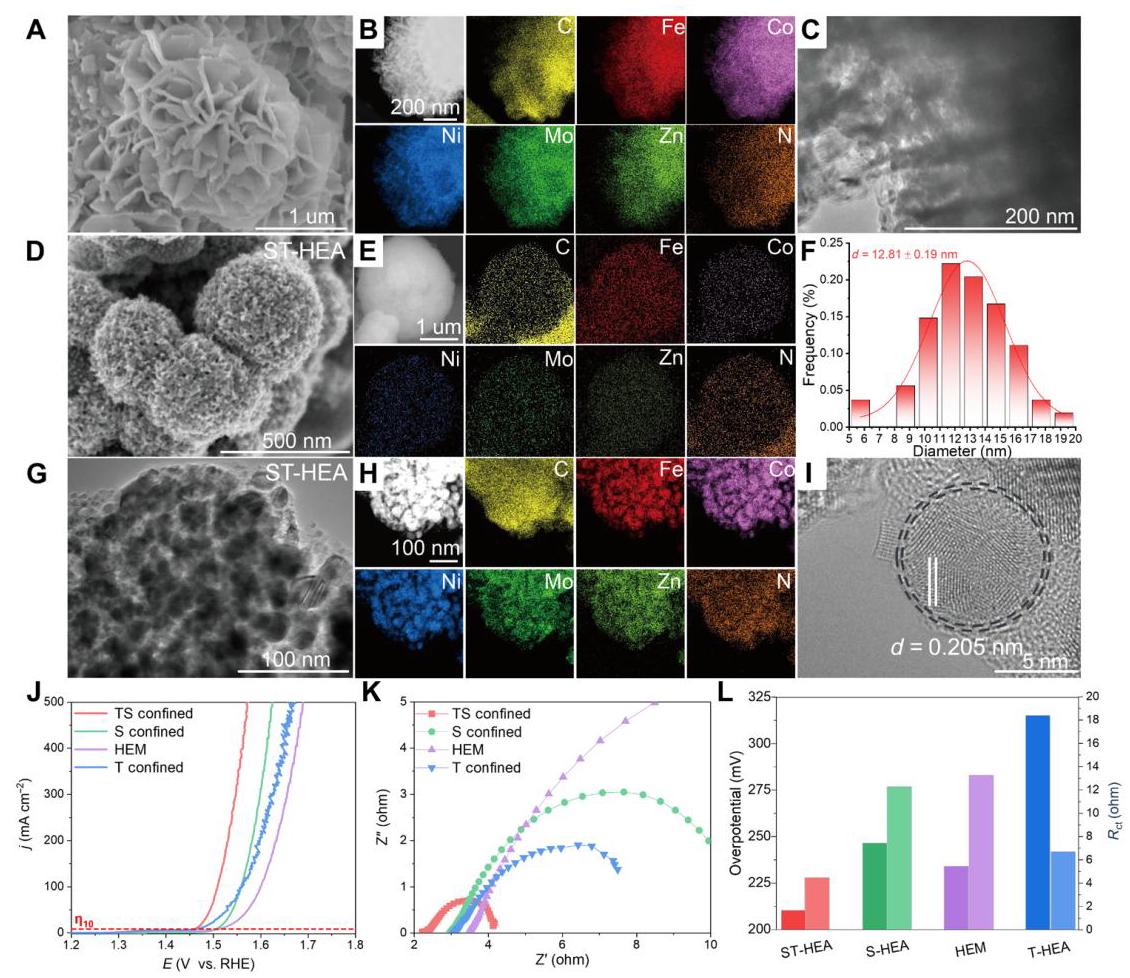
图3. MoZn基HEA的详细表征。(A) 高熵MOF的SEM图像和(B) Fe、Co、Ni、Mo、Zn、C和N元素均匀分布的EDS图谱。(C) 高熵MOF的TEM图像。(D) ST-HEA的SEM图像。(E) Fe、Co、Ni、Mo、Zn、C和N元素均匀分布的EDS图谱。(F) ST-HEA的粒径分布。(G) 单个多层ST-HEA的TEM。(H) Fe、Co、Ni、Mo、Zn、C和N元素均匀分布的EDS图谱。(I) HEA的HRTEM图像显示HEA被碳壳层包裹。(J) OER极化曲线,(K) EIS奈奎斯特图和(L) ST-HEA、S-HEA、高熵MOF和T-HEA催化剂在10 mA cm⁻²下的过电位和电荷转移电阻(Rct)比较。
分析结果
图3A-C显示高熵MOF前驱体具有花状结构,由交错重叠的片层组成,具有大表面积。EDS图谱证实C、Fe、Co、Ni、Mo和Zn六种元素在选定颗粒区域内均匀分布。图3D-E显示ST-HEA形成高度多孔的球形结构,由碳框架连接的纳米颗粒自组装而成,五种元素在高温热解后均匀分散无元素偏析。TEM分析(图3F-I)显示ST-HEA具有均匀的纳米颗粒形态(尺寸为12.81±0.19 nm),由碳框架互连,晶格条纹为0.205 nm,归属于HEA的(111)晶面,且合金颗粒被源自MOF模板的薄碳壳无缝覆盖。电化学测试(图3J-L)表明,ST-HEA具有最佳的OER活性(221 mV@10 mA cm⁻²)和最低的电荷转移电阻(2.5 ohm),这归因于分级多孔结构和石墨化碳壳。
图4: 制备催化剂和商业催化剂的电催化性能
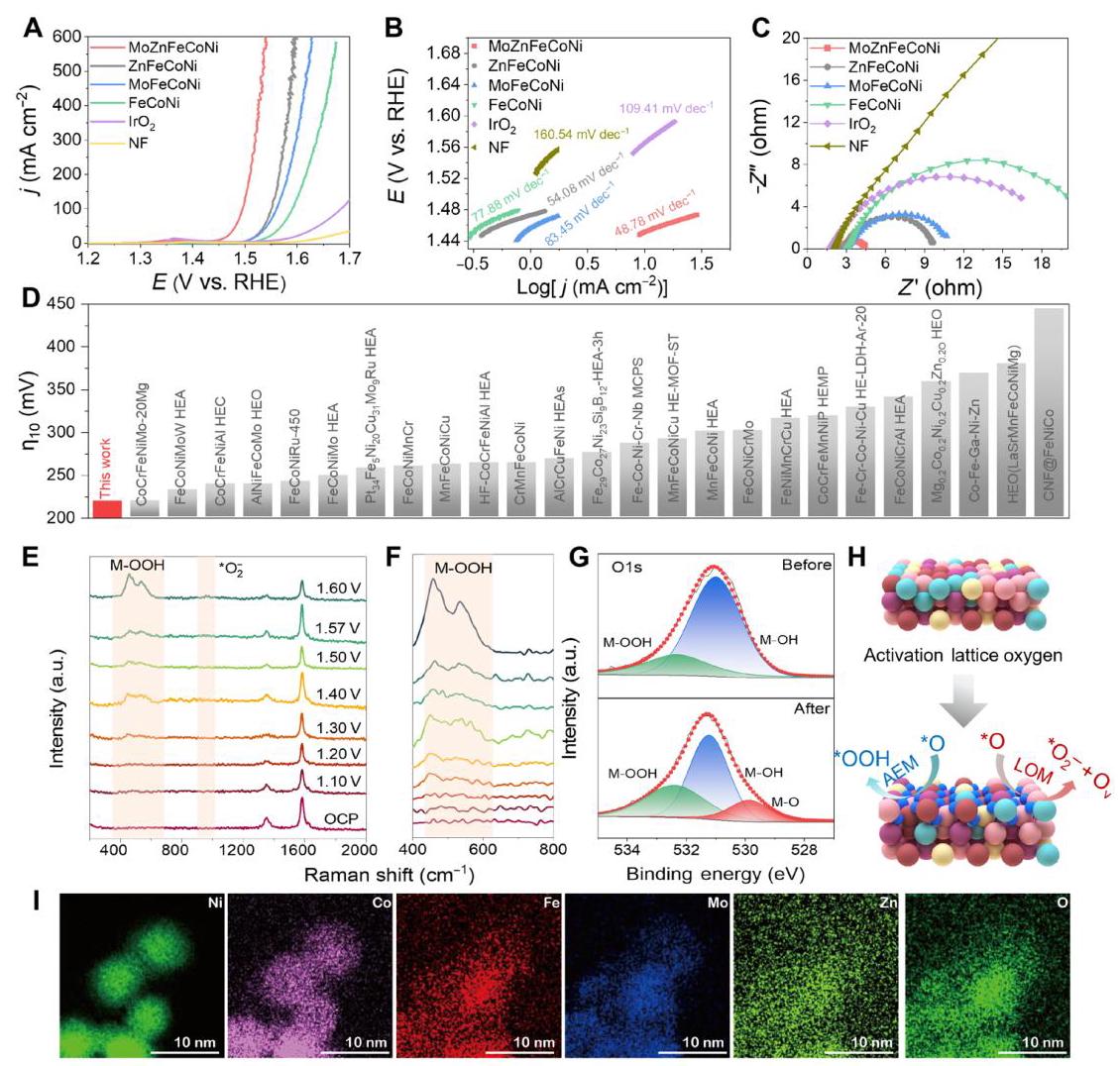
图4. 制备催化剂和商业催化剂的电催化性能。(A) 在10 mA cm⁻²下的IR校正极化曲线和(B) MoZnFeCoNi、ZnFeCoNi、MoFeCoNi和FeCoNi催化剂在1.0 M KOH溶液中的塔菲尔图。(C) MoZnFeCoNi、IrO₂和NF的EIS奈奎斯特图。(D) MoZnFeCoNi催化剂在10 mA cm⁻²下的过电位与近期报道的高熵OER电催化剂在1.0 M KOH中的比较。(E-F) 在1 M KOH中收集的MoZnFeCoNi上OER的原位拉曼光谱。(G) OER活化前后催化剂O 1s的XPS分析。(H) OER活化后MoZnFeCoNi复合物表面重构和OER机制双重激活的示意图。(I) MoZnFeCoNi的相应EDX图谱图像。
分析结果
图4A-B显示MoZnFeCoNi催化剂在10 mA cm⁻²下具有最低的过电位(221 mV)和塔菲尔斜率(48.78 mV dec⁻¹),表明其具有更快的OER动力学。图4C的EIS分析表明MoZnFeCoNi具有最低的电荷转移电阻(2.5 ohm),远低于商业IrO₂(18.3 ohm)。图4D显示MoZnFeCoNi的OER活性优于许多近期报道的高熵电催化剂。原位拉曼光谱(图4E-F)显示在1.2-1.4 V vs. RHE电压范围内出现473.0和552.4 cm⁻¹的M-O振动峰,表明催化剂在OER过程中发生结构重构,同时观察到含氧中间体和*O₂⁻物种,表明AEM和LOM双重激活。XPS分析(图4G)显示活化后催化剂中存在晶格氧(M-O)、缺陷氧(M-OH)和吸附氧物种(M-OOH),金属逐渐氧化生成MOOH,被认为是OER的真实催化物种。
图5: OER过程中的DFT计算和机理分析
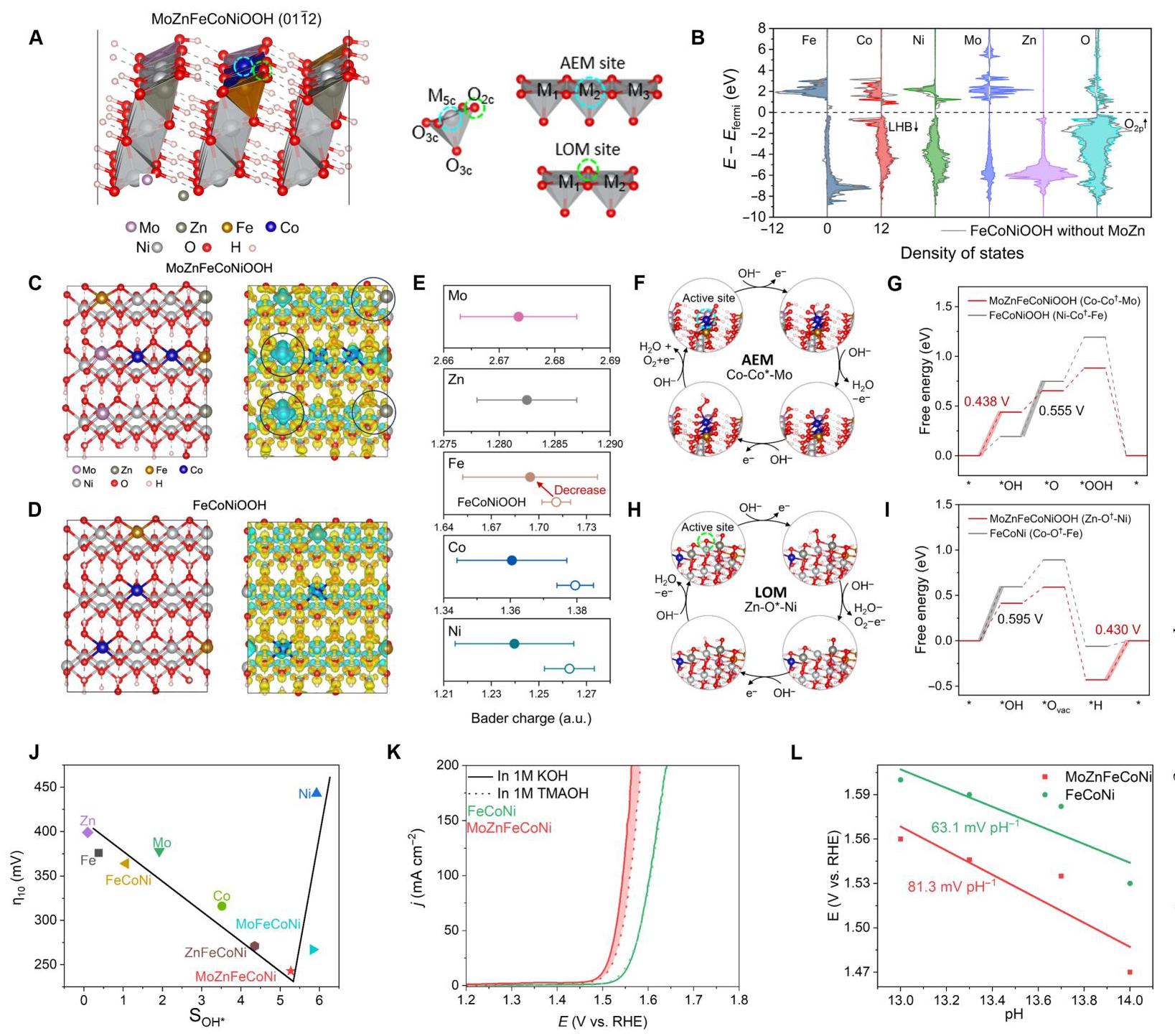
图5. OER过程中的DFT计算和机理分析。(A) 用于DFT计算的MoZnFeCoNiOOH(01ī2)面模型。蓝色和绿色圆圈表示AEM和LOM路径的活性位点。标记了金属和氧原子的配位,其中5c、3c和2c代表五重、三重和二重配位。M₁-M₂*-M₃和M₁-O*-M₂是对AEM和LOM性能至关重要的排列。(B) MoZnFeCoNiOOH模型中表面Fe、Co、Ni、Mo、Zn和O原子的部分态密度(PDOS),其中FeCoNiOOH的结果显示为灰线。(C-D) MoZnFeCoNiOOH和FeCoNiOOH的平板模型和电荷密度差。黄色和蓝色分别代表电子积累和耗尽,等值面值为0.02 eÅ⁻³。(E) MoZnFeCoNiOOH(实心点)和FeCoNiOOH(空心点)外表面位点的Bader电荷统计分析。点和误差条显示Bader电荷的平均值和标准差。(F-G) Co-Co*-Mo位点的AEM路径和相应的OER步骤吉布斯自由能图。自由能是在1.23 V vs. RHE下获得的。为了比较,给出了FeCoNiOOH的自由能图。(H-I) Zn-O*-Ni位点的LOM路径和相应的OER步骤吉布斯自由能图。(J) MOR中过电位与导致催化剂中填充区域的电流差之间的关系。(K) FeCoNi和MoZnFeCoNi分别在1.0 M KOH和1.0 M TMAOH中的LSV曲线。(L) MoZnFeCoNi和FeCoNi在1.5 V vs. RHE电位下的电流密度对数与pH的关系。
分析结果
DFT计算揭示了MoZnFeCoNiOOH的OER机理。图5B的PDOS分析显示,添加Mo和Zn后,整体LHB中心从-4.342下移至-4.794 eV,而O-2p带从-2.480上移至-2.210 eV,减少了金属d轨道和O-2p轨道的重叠,削弱了金属-氧键,可触发LOM机制。图5C-E的电荷密度差和Bader电荷分析显示,Mo原子周围有明显的电荷耗尽,Zn采用低价Zn²⁺态,导致沿Zn-O键的O原子周围电子局域化,削弱了键强度。AEM路径中(图5F-G),Co-Co*-Mo位点表现出最低的过电位(0.438 V),PDS为*OH到*O的转化。LOM路径中(图5H-I),Zn-O*-Ni位点表现出最优的过电位(0.430 V),PDS为羟基攻击步骤。实验验证(图5J-L)通过甲醇氧化反应(MOR)和四甲基铵阳离子(TMA⁺)分子探针证实了双重机制激活,MoZnFeCoNi表现出最高的OER活性和适中的*OH吸附,且OER活性高度依赖于电解质pH,进一步表明LOM的存在。
图6: MoZnFeCoNi的电催化OER稳定性
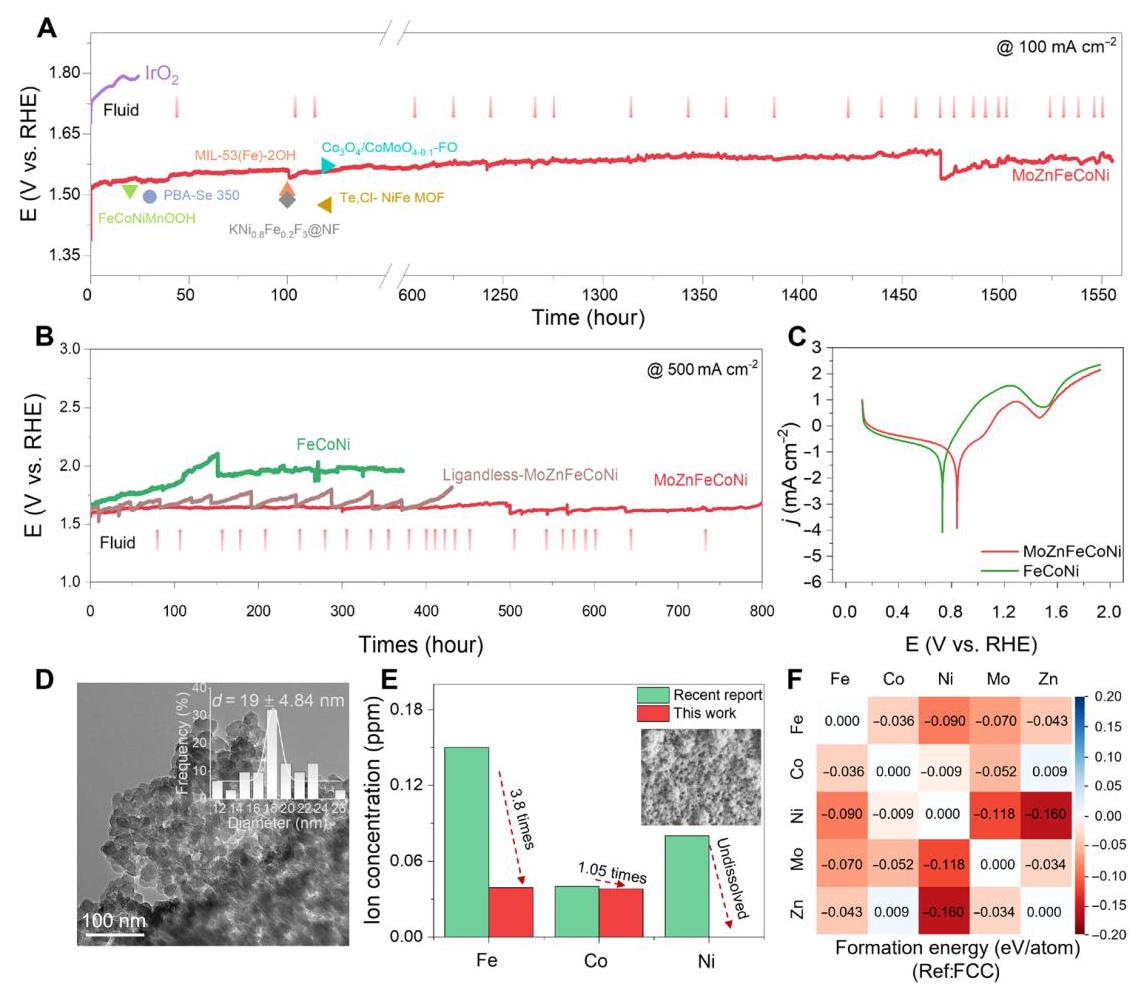
图6. MoZnFeCoNi的电催化OER稳定性。(A) 在1.0 M KOH中100 mA cm⁻²下的计时电位法测量催化稳定性。(B) 在500 mA cm⁻²下的稳定性。(C) 催化剂的腐蚀电位分析。(D) 在500 mA cm⁻²下稳定性测试后MoZnFeCoNi的TEM图像(插图为分级MoZnFeCoNi中单个颗粒的粒径分布)。(E) 在100 mA cm⁻²恒定电流密度下稳定性测试后电解质中的相应离子浓度。(插图SEM为强酸蚀刻后MoZnFeCoNi的碳网络)。(F) MoZnFeCoNi中元素对之间相对于组成元素FCC结构的形成能。
分析结果
图6A-B显示MoZnFeCoNi在100 mA cm⁻²下表现出超过1500小时的持续催化活性,在500 mA cm⁻²下经过800小时稳定性测试后电位几乎不变,显著优于商业IrO₂(仅持续24小时)和大多数已报道催化剂。图6C的动电位极化(PDP)曲线表明,MoZnFeCoNi具有较高的腐蚀电位(0.83 V vs. RHE),优于FeCoNi(0.73 V vs. RHE),表明其在碱性电解液中具有优异的抗腐蚀能力。稳定性测试后的TEM和EDX结果显示(图6D),分级结构保持良好,Fe、Co、Ni、Mo、Zn、C、N和O元素仍然均匀分布在整个催化剂区域,但单个颗粒尺寸明显增大,可能是由于电化学重构后的表面氧化所致。ICP-OES分析(图6E)显示,经过1500小时稳定性测试后,Fe/Co/Ni/Mo的溶解量极低,Zn虽然溶解度较高,但平均溶解速率仅为1.27 ppb/小时,确保了OER耐久性。DFT计算的形成能分析(图6F)显示,Mo和Zn与几乎所有其他元素都显示负结合能,表明多元素相互作用和高熵稳定在提高结构稳定性方面发挥重要作用。