Phase controlled synthesis of transition metal carbide nanocrystals by ultrafast flash Joule heating
超快闪烧焦耳加热法相控合成过渡金属碳化物纳米晶体
Bing Deng1, Zhe Wang1, Weiyin Chen1, John Tianci Li1,2, Duy Xuan Luong1, Robert A. Carter1, Guanhui Gao2, Boris I. Yakobson1,2,3, Yufeng Zhao2,4∞ & James M. Tour1,2,3,5∞
1 Department of Chemistry, Rice University, Houston, TX 77005, USA.
2 Department of Materials Science and NanoEngineering, Rice University, Houston, TX 77005, USA.
3 Smalley-Curl Institute, Rice University, Houston, TX 77005, USA.
4 Corban University, Salem, Oregon 97317, USA.
5 NanoCarbon Center and the Welch Institute for Advanced Materials, Rice University, Houston, TX 77005, USA.
DOI: 10.1038/s41467-021-27878-1
PDF原文
期刊: Nature Communications
发表年份: 2022
论文亮点
- 开发了一种超快闪烧焦耳加热(FJH)方法,可在1秒内合成无焦炭、相控的碳化物纳米晶体,克服了传统方法耗时、易结焦、粒径大和相不可控的缺点。
- 通过调控脉冲电压,成功实现了对钼碳化物(包括热力学稳定的β-Mo₂C和亚稳态的α-MoC₁₋ₓ、η-MoC₁₋ₓ)的相选择性合成,并发现β-Mo₂C相具有最优的析氢反应(HER)性能。
研究背景
- 过渡金属碳化物(TMCs)因其极高的硬度、高热稳定性和可广泛调控的电子结构,在电子学、陶瓷和能源转换等领域有广泛应用。纳米尺寸的TMCs是超硬超强陶瓷的前驱体,因其类铂的电子结构而成为高性能电化学催化剂,并因强烈的金属-载体相互作用而作为催化剂载体。
- 传统的块状碳化物合成方法(如金属前驱体与气态碳源渗碳或在高温下与石墨碳烧结)通常存在问题:碳源过量供应导致碳化物表面结焦,粒径大且比表面积低,不利于催化性能。现有的其他方法(如程序升温还原、碳热还原、激光喷雾热解、溶液沉淀与渗碳)也存在反应窗口需优化、耗时、使用昂贵有毒前体、冷却速度慢无法保留亚稳相等局限性。
- 相的调控对碳化物的性能至关重要(如氢吸附/脱附能),但目前很少有方法能够选择性地调控碳化物的相和晶体表面以获得最佳性能。
研究方法
超快闪烧焦耳加热(FJH)合成:
- 前驱体准备: 将金属前驱体(金属单质、氧化物、氯化物、氢氧化物等)与商业炭黑(既作碳源也作导电添加剂)按特定重量比(补充表2)在研钵中研磨混合。
- 反应装置: 将约50 mg反应物装入石英管(内径4 mm,外径8 mm)中,石英管两端用石墨棒作为电极。电极与石英管松散配合以允许排气。
- 反应过程: 将装载样品的反应台放入密封反应室,抽至轻度真空(~10 mmHg)以容纳脱气并避免样品氧化。使用总电容为60 mF的电容器组作为电源。通过可编程ms级延迟时间的继电器控制放电时间。充电、闪烧加热和放电过程通过National Instruments Multifunction I/O (NI USB-6009) 结合定制LabView程序自动控制。
- 能量输入与温度: 一个毫秒级的电流脉冲通过前驱体,使样品在极短时间内达到超高温(>3000 K),随后以超快速度冷却至室温(>10⁴ K s⁻¹)。通过调节放电电压和/或电容,可在不同质量规模上获得恒定的温度值和均匀性。
- 相控制: 通过控制FJH脉冲电压,可以选择性合成不同相的碳化物(例如对于钼碳化物,30 V得β-Mo₂C,60 V得α-MoC₁₋ₓ,120 V得η-MoC₁₋ₓ)。
- 后续处理(可选): 根据应用需求,可通过空气中简单煅烧(用于SiC)、Ca金属蚀刻(用于TiC, ZrC, HfC, VC, NbC, TaC, Cr₃C₂, β-Mo₂C, W₂C)或液体内密度纯化程序(用于亚稳态钼碳化物α-MoC₁₋ₓ和η-MoC₁₋ₓ)来去除过量碳(石墨烯支撑体)以实现碳化物的高效纯化。
表征方法: 使用SEM/EDS、Raman、XRD、XPS、BF-TEM/HRTEM、STEM/HAADF/EDS、BET等方法对合成的碳化物纳米晶进行形貌、成分、晶体结构、电子结构、尺寸等的表征。
理论计算: 采用基于第一性原理的密度泛函理论(DFT)计算(通过VASP软件包实现)研究不同碳含量下碳化物的形成能、相变路径以及氢吸附自由能(ΔGH),以解释实验现象。
电化学测试: 使用标准三电极体系在0.5 M H₂SO₄溶液中测量不同相钼碳化物的析氢反应(HER)性能,包括线性扫描伏安法(LSV)、塔菲尔斜率(Tafel slope)、电化学阻抗谱(EIS)和循环伏安法(CV)耐久性测试。
主要结论
- 成功开发了超快闪烧焦耳加热(FJH)方法,可在1秒内通用地合成多种无焦炭、相控的碳化物纳米晶体,包括间隙式TMCs(TiC, ZrC, HfC, VC, NbC, TaC, Cr₂C₃, MoC, W₂C)和共价碳化物(B₄C, SiC)。
- 通过精确控制FJH脉冲电压,实现了对钼碳化物相的选择性合成(β-Mo₂C, α-MoC₁₋ₓ, η-MoC₁₋ₓ),证明了FJH过程通过广泛可调的能量输入和动力学控制的超快冷却速率所具有的优异相工程能力。DFT计算揭示碳空位是碳化物相拓扑转变的驱动因素。
- 发现了钼碳化物的相依赖析氢反应(HER)性能,其中β-Mo₂C表现出最佳的HER性能(过电位-220 mV,塔菲尔斜率68 mV dec⁻¹)和良好的耐久性。DFT计算表明其优异的性能源于相对较小的氢吸附能、增强的金属特性和较高的比表面积。
结果与分析:超快闪烧焦耳加热合成碳化物纳米晶体
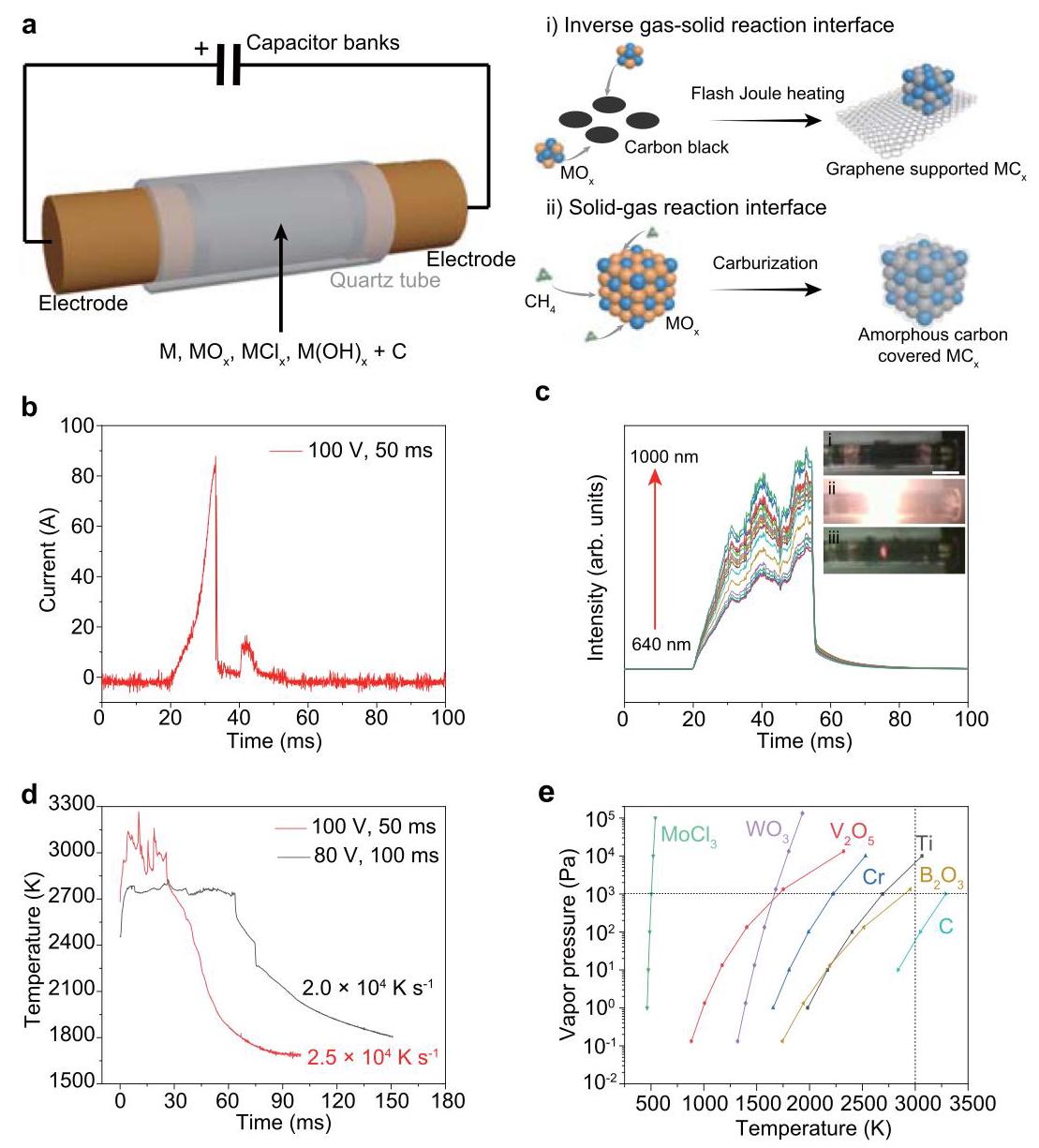
图1:超快闪烧焦耳加热(FJH)合成碳化物示意图及相关测量。(a) FJH合成碳化物示意图。路线(i)本文披露的高温FJH过程。路线(ii)传统的渗碳过程。(b) FJH过程中的电流测量。(c) 640-1000 nm波长范围内的实时光谱辐射亮度。插图:FJH前(i)、FJH过程中(ii)和快速冷却(iii)的样品图片。(d) 通过拟合FJH过程中样品的黑体辐射进行实时温度测量。(e) 各种金属前驱体和碳的温度-蒸气压关系。
分析结果: FJH过程通过毫秒级放电将样品迅速加热至超高温(~3000 K),随后超快冷却(>10⁴ K s⁻¹)。温度通过黑体辐射光谱拟合测量。温度分布模拟(FEM)显示整个样品温度均匀。根据温度-蒸气压关系,金属前驱体为挥发性组分,碳源在反应过程中保持固态,形成了金属前驱体蒸气与碳反应生成金属碳化的“反向气-固反应界面”,这与传统渗碳过程(气态碳源与固态金属反应,易导致表面结焦)相反,从而避免了表面结焦问题。
结果与分析:相控合成钼碳化物纳米晶体
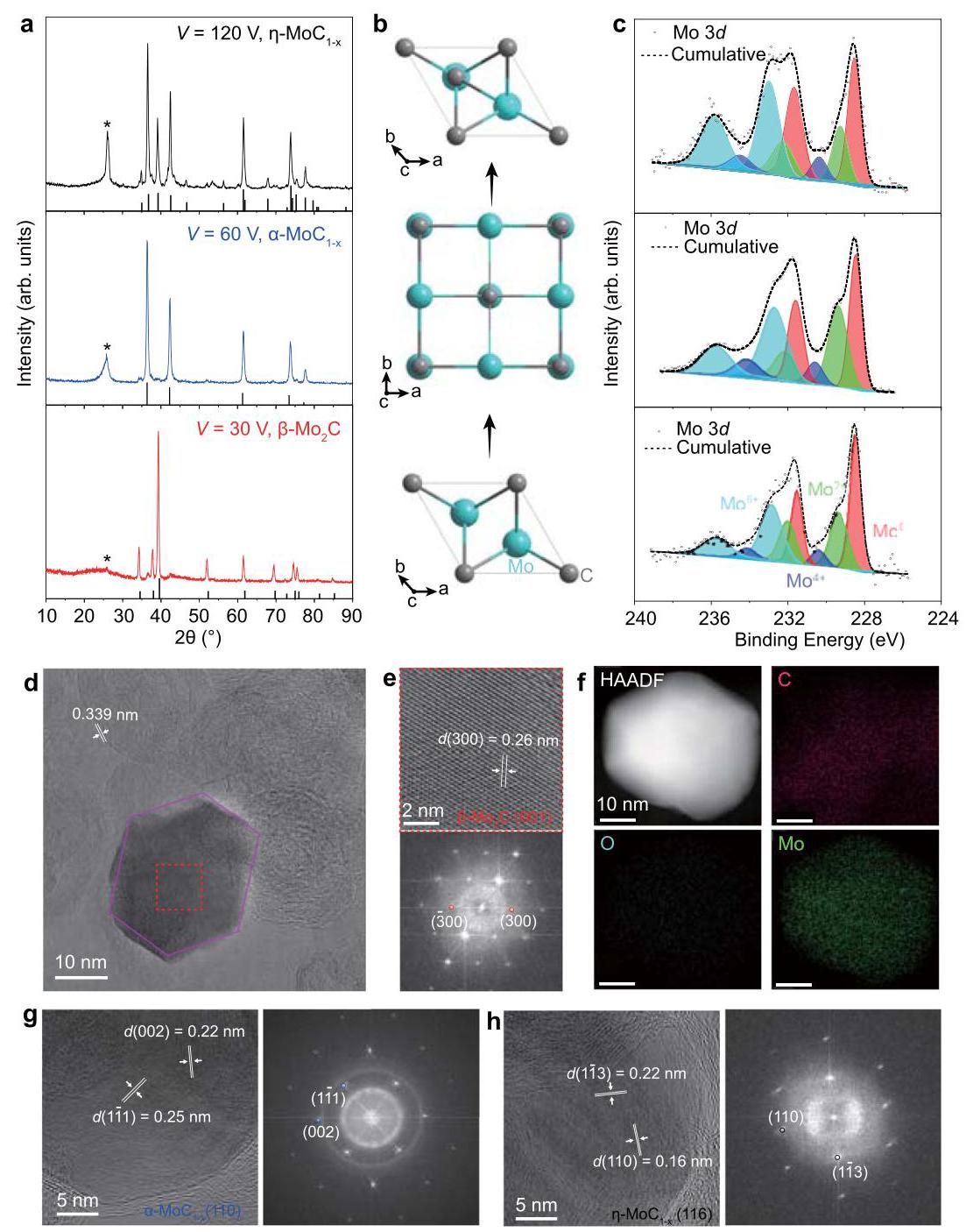
图2:相控合成钼碳化物。(a) 分别在30 V, 60 V, 120 V电压下合成的β-Mo₂C, α-MoC₁₋ₓ, η-MoC₁₋ₓ的X射线衍射(XRD)图。(b) 三种钼碳化物相的晶体结构。(c) 三种钼碳化物相的X射线光电子能谱(XPS)谱图(Mo 3d核心能级)。(d) 支撑在石墨烯上的β-Mo₂C纳米晶的明场透射电子显微镜(BF-TEM)图像。(e) β-Mo₂C的高分辨率透射电子显微镜(HRTEM)图像(上)和相应的快速傅里叶变换(FFT)图(下)。(f) β-Mo₂C的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像和能量色散谱(EDS)元素分布图。(g) α-MoC₁₋ₓ的HRTEM图像(左)和相应的FFT图(右)。(h) η-MoC₁₋ₓ的HRTEM图像(左)和相应的FFT图(右)。
分析结果: 通过调节FJH电压,成功选择性合成了三种纯相的钼碳化物:30 V得到六方β-Mo₂C,60 V得到立方α-MoC₁₋ₓ,120 V得到六方η-MoC₁₋ₓ。XPS分析表明β-Mo₂C具有最好的抗氧化性。TEM和XRD显示合成的碳化物纳米颗粒多为单晶,粒径受电压调控(较高电压下粒径更小,归因于更高温度下更快的成核动力学)。HRTEM和FFT证实了各相的晶体结构和取向。这是一种新发现的从β-Mo₂C到α-MoC₁₋ₓ再到η-MoC₁₋ₓ的拓扑转变路径。
结果与分析:DFT计算揭示钼碳化物相变过程
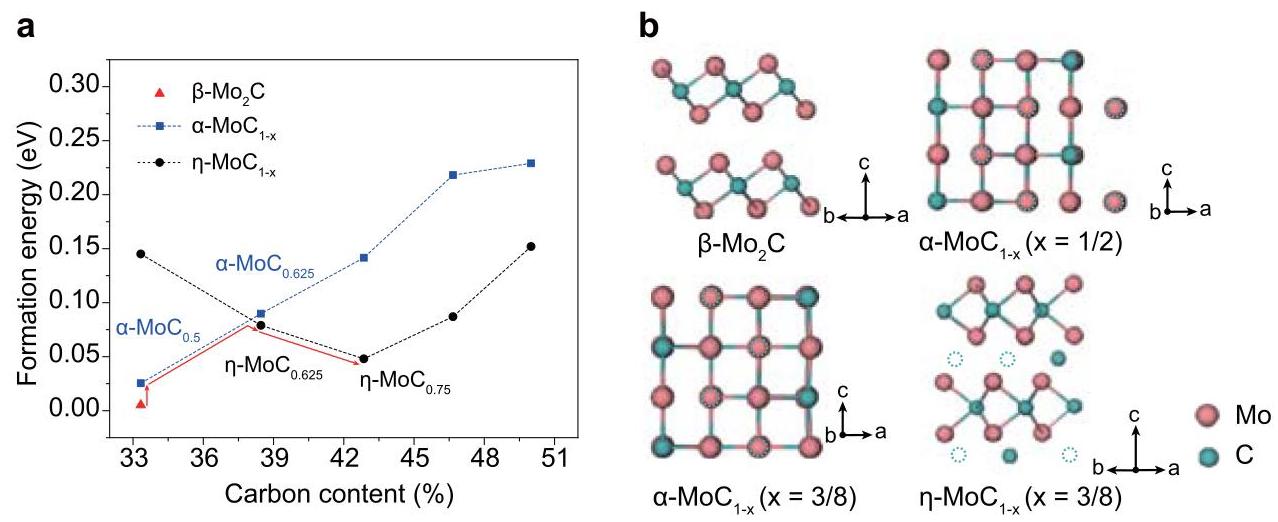
图3:密度泛函理论(DFT)计算揭示的钼碳化物相变过程。(a) β-Mo₂C、α-MoC₁₋ₓ和η-MoC₁₋ₓ随碳原子含量变化的形成能。红线表示预测的相变路径。(b) 计算得到的β-Mo₂C、α-MoC₁₋ₓ (x=1/2)、α-MoC₁₋ₓ (x=3/8) 和 η-MoC₁₋ₓ (x=3/8) 的晶体结构。虚线圆圈表示碳空位。
分析结果: DFT计算表明,β-Mo₂C相是形成能最低的最稳定相,因此在相对较低的电压和温度下形成。α-MoC₁₋ₓ和η-MoC₁₋ₓ是亚稳态相,在更高温度下形成和稳定。碳空位主导了Mo-C体系的能量分布,是从β-Mo₂C到α-MoC₁₋ₓ再到η-MoC₁₋ₓ相拓扑转变的驱动因素。FJH过程广泛可调的能量输入允许获得形成能高于热力学稳定相的亚稳态相,而其超快冷却速率(>10⁴ K s⁻¹)有助于动力学地将亚稳态相(α-MoC₁₋ₓ和η-MoC₁₋ₓ)保留至室温。对照实验(使用传统管式炉慢速冷却)在相同温度下只能得到热力学稳定的β-Mo₂C相,证明了FJH超快冷却速率在动力学上获得亚稳态相的关键作用。
结果与分析:钼碳化物的相依赖HER性能
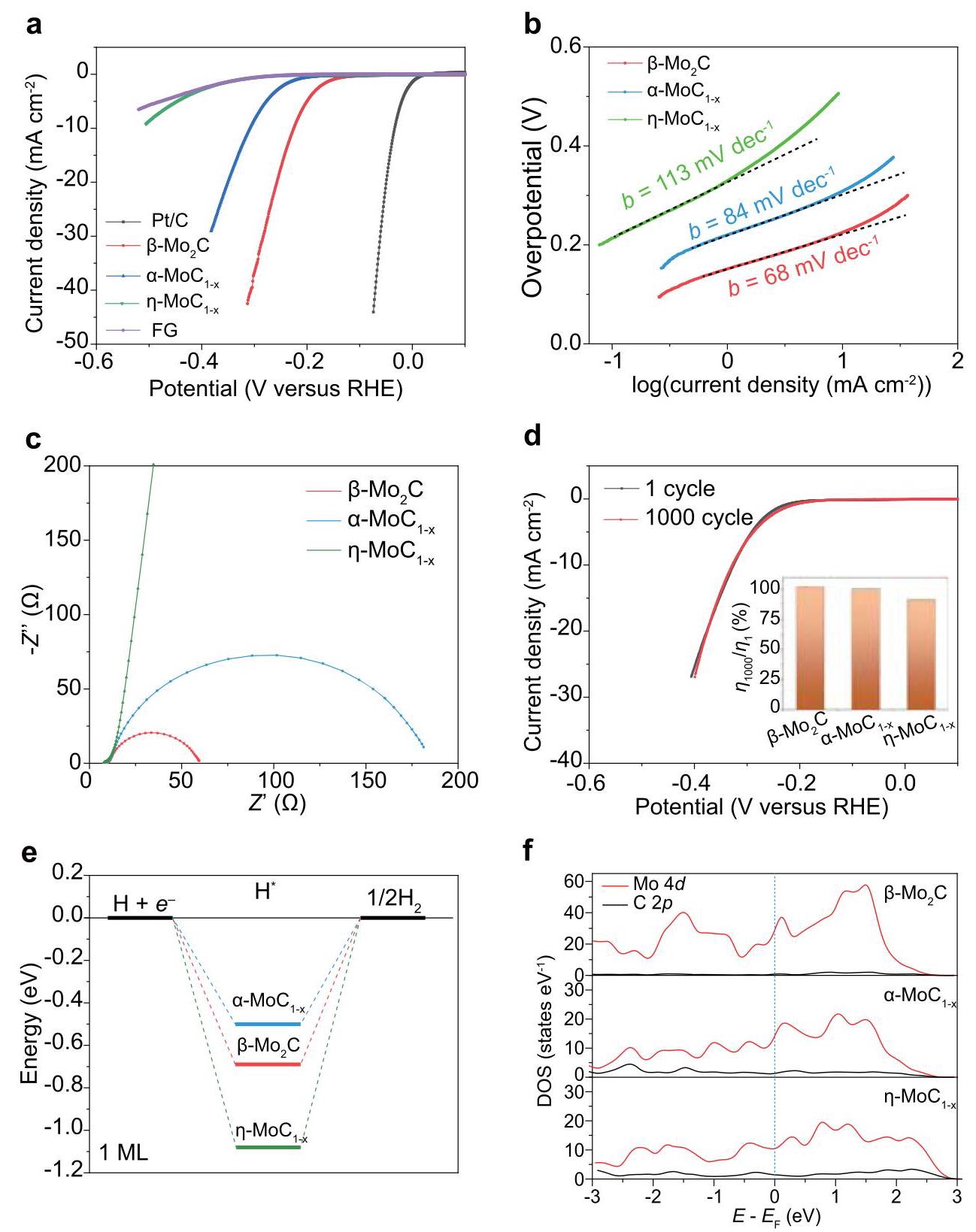
图4:钼碳化物的相依赖析氢反应(HER)性能。(a) 三种钼碳化物相的极化曲线。Pt/C和纯闪烧石墨烯(FG)用作对照。(b) 三种钼碳化物相的塔菲尔曲线。(c) 三种钼碳化物相的交流(AC)阻抗。(d) 钼碳化物的耐久性,显示α-MoC₁₋ₓ第1次和第1000次循环的极化曲线。插图为三种钼碳化物相第1次和第1000次循环的过电位比率。(e) 在单层氢吸附覆盖度下,β-Mo₂C(001)、α-MoC₁₋ₓ(110)和η-MoC₁₋ₓ(001)上HER的自由能图。(f) β-Mo₂C (001)、α-MoC₁₋ₓ (110)和η-MoC₁₋ₓ (001)中Mo和C的计算部分态密度。蓝色虚线表示费米能级位置。
分析结果: 电化学测试表明钼碳化物的HER性能具有相依赖性。β-Mo₂C表现出最佳的HER性能(η₁₀ = -220 mV,Tafel斜率 = 68 mV dec⁻¹,电荷转移电阻~60 Ω),其次是α-MoC₁₋ₓ(η₁₀ = -310 mV),η-MoC₁₋ₓ性能最差(η₁₀ = -510 mV)。三种相均表现出良好的长期稳定性(1000次循环后性能衰减很小)。DFT计算表明,β-Mo₂C(001)的氢吸附自由能(ΔGH = 0.48 eV)比α-MoC₁₋ₓ(110)(0.71 eV)和η-MoC₁₋ₓ(001)(1.09 eV)更接近理想值(0 eV)。此外,β-Mo₂C在费米能级附近的态密度(DOS)更大,表明其更高的载流子密度和增强的金属特性,有利于电化学反应过程中的电荷转移。BET测量显示β-Mo₂C具有更大的比表面积。β-Mo₂C最佳的HER性能是其相对较小的氢吸附能、增强的金属特性和高比表面积共同作用的结果。闪烧石墨烯支撑体提供了导电通路并防止碳化物纳米晶聚集,也有助于提高HER性能。
结果与分析:碳化物纳米晶体合成的通用策略
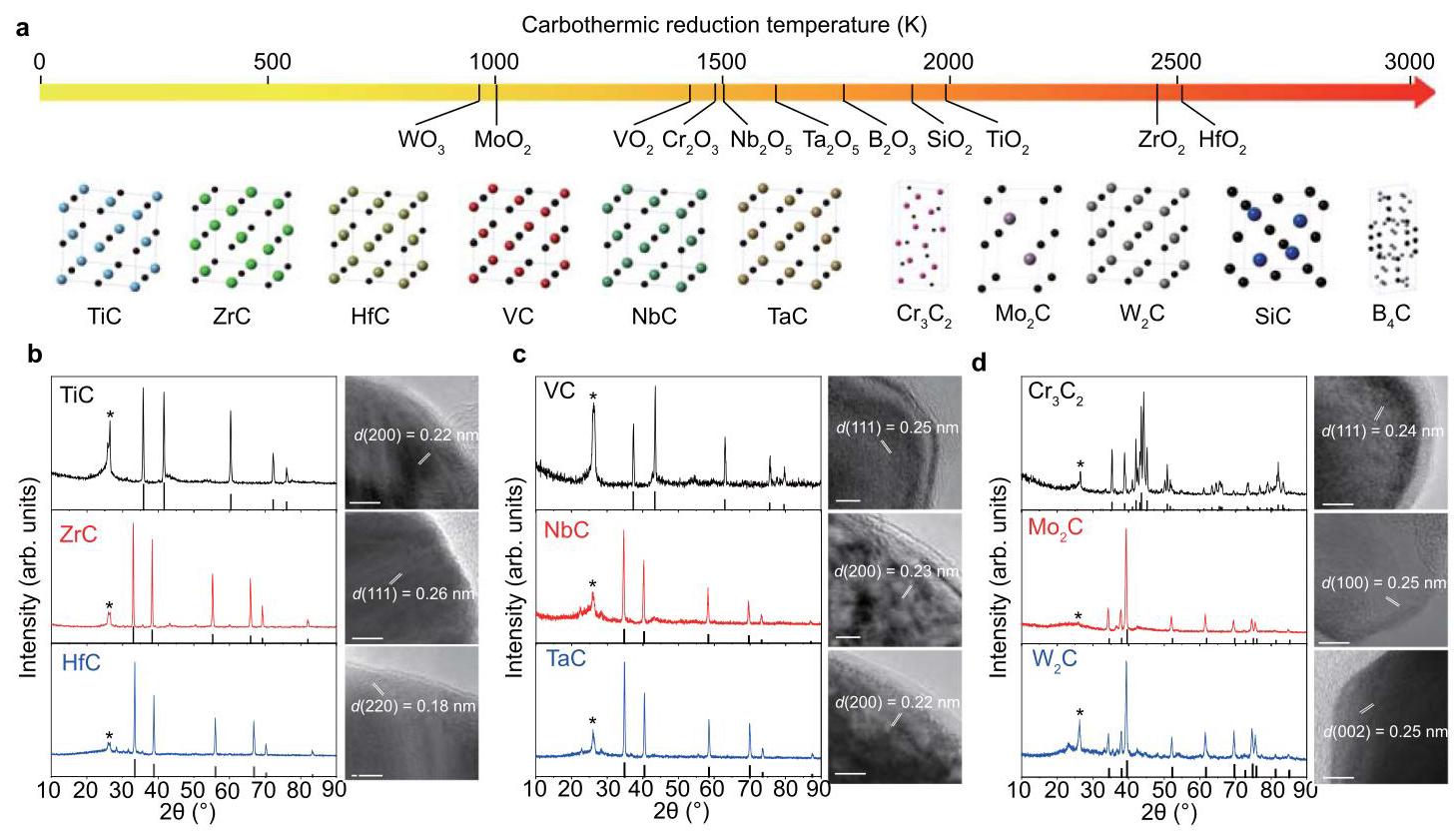
图5:碳化物合成的通用策略。(a) 由Ellingham图推导的氧化物碳热还原温度,以及11种碳化物的晶体结构。(b) IVB族金属碳化物的X射线衍射(XRD)图和高分辨率透射电子显微镜(HRTEM)图像。(c) VB族金属碳化物的XRD图和HRTEM图像。(d) VIB族金属碳化物的XRD图和HRTEM图像。
分析结果: 利用FJH过程的超高温(~3000 K),成功合成了来自IVB、VB、VIB过渡族的多种碳化物纳米晶体(TiC, ZrC, HfC, VC, NbC, TaC, Cr₃C₂, Mo₂C, W₂C)以及共价碳化物B₄C和SiC,证明了FJH方法的普适性。均匀的温度分布保证了整个样品中相纯的合成。使用低成本金属或其化合物(氧化物、氢氧化物、氯化物)作为前驱体,使得FJH与依赖挥发性化合物的先前方法相比成为一种有前景的低成本生产方法。对于VIB族碳化物(Cr, Mo, W),相组成更为复杂。成功合成了亚稳态的W₂C相(根据W-C相 diagram,其在1250°C以下不如WC相稳定),这再次证明了FJH过程通过高能量输入和超快冷却速率所具有的优异相工程能力。合成的碳化物纳米颗粒多为单晶(XRD与TEM尺寸测量结果吻合)。