Tailoring the Local Environment of Platinum in Single-Atom Pt1/CeO2 Catalysts for Robust Low-Temperature CO Oxidation
单原子催化:调控Pt1/CeO2催化剂的局部环境以实现稳健的低温CO氧化
Dong Jiang, Yonggang Yao, Tangyuan Li, Gang Wan, Xavier Isidro Pereira-Hernández, Yubing Lu, Jinshu Tian, Konstantin Khivantsev, Mark H. Engelhard, Chengjun Sun, Carlos E. García-Vargas, Adam S. Hoffman, Simon R. Bare, Abhaya K. Datye*, Liangbing Hu*, and Yong Wang*
DOI: 10.1002/anie.202108585 | Angewandte Chemie International Edition | 2021
PDF原文
论文亮点
- 开发了一种热冲击(TS)合成方法,成功制备了具有不对称Pt1O4配位结构的单原子Pt1/CeO2催化剂。
- 该催化剂在低温CO氧化反应中表现出优异的活性和稳定性,且在氧化条件下活性保持良好,解决了传统原子捕获(AT)方法制备的催化剂活性低或稳定性差的问题。
研究背景
- 单原子催化剂(SACs)在最大化原子利用率和提高催化活性/选择性方面展现出巨大潜力,但同时实现高反应活性和高热稳定性是其在工业应用中面临的主要挑战。
- CO氧化在汽车尾气处理中至关重要,需要兼具高活性和高热稳定性的催化剂。传统的原子捕获(AT)方法(空气中800°C热处理)可制备热稳定的Pt1/CeO2催化剂,但其Pt2+处于高度对称的平面四方Pt1O4配位环境中,过度稳定,导致低温CO氧化活性几乎为零。
- 通过还原活化形成Pt纳米颗粒(NPs)可增强活性,但NPs在氧化条件下易被氧化,导致活性急剧下降,这限制了其在排放控制系统(尤其是发动机冷启动)中的应用。
研究方法
本研究采用两种高温合成方法制备了1 wt% Pt负载的Pt1/CeO2催化剂:
- 原子捕获法(AT): 将四氨硝酸铂(TAPN)前驱体通过初湿浸渍法(IWI)负载到CeO2上,随后在空气中800°C下长时间热处理。此方法使Pt原子被捕获在CeO2最稳定的位点(如单原子台阶边缘),形成对称的平面四方Pt1O4配位结构。
- 热冲击法(TS): 使用相同的TAPN/CeO2前驱体,在惰性气氛中进行超高温热冲击处理(>1200°C)。该过程由短暂的“开启”状态(约500 ms)和较长(约6倍)的“关闭”状态组成周期性加热。超高温闪烧通过重构CeO2表面驱动Pt分散并形成强Pt-O-Ce键,而快速冷却(约104 K s-1)则防止Pt和CeO2的烧结。惰性气氛限制了PtO2的气相传输,使Pt原子稳定在不同于AT方法的最稳定位点上,最终形成具有不对称Pt1O4配位结构的Pt2+单原子。
采用多种表征技术对催化剂进行系统分析:
- AC-STEM: 确认Pt的原子级分散。
- XAS(XANES和EXAFS): 分析Pt的氧化态和局部配位环境。
- XPS: 分析表面元素组成和化学状态。
- BET: 测量比表面积。
- 原位DRIFTS: 以CO为探针分子,研究反应条件下Pt物种的性质和演变。
- 催化性能测试: 在富氧条件下评价催化剂的CO氧化活性( light-off 曲线)和稳定性(氧化/还原处理后的循环测试)。
- 动力学研究: 测量表观活化能和反应级数。
主要结论
- 热冲击(TS)法成功制备了具有不对称Pt1O4配位结构的Pt1/CeO2单原子催化剂,而原子捕获(AT)法则得到高度对称的Pt1O4配位结构。
- TS催化剂表现出显著增强的低温CO氧化活性(T50 ~150°C),远优于AT催化剂(T50 ~287°C),且活性在重复起燃测试中保持稳定。
- TS催化剂在500°C氧化处理后仅表现出微小的活性衰减,展现出优异的抗氧化失活能力;而AT催化剂经还原活化形成Pt NPs后虽活性提高,但在高温氧化后活性会急剧下降至初始水平。
- 不对称配位环境使得Pt2+在CO氧化反应条件下能动态演变为部分还原的Pt1δ+物种(Pt1O4-x配位),这些物种是低温高活性的关键,且该过程是可逆的,避免了形成易氧化的金属态Pt NPs。
图表分析:催化剂合成与结构表征
图1a展示了AT和TS两种合成路线的示意图及其导致的Pt单原子局部配位环境差异。
分析结果: AT法产生高度对称的(近乎完美的)平面四方Pt1O4配位,而TS法由于超高温引起的CeO2表面重构和受限的PtO2气相传输,形成了不对称的(扭曲的平面四方)Pt1O4配位。
图1b和1c的AC-STEM图像证实了两种催化剂中Pt均以原子形式分散。
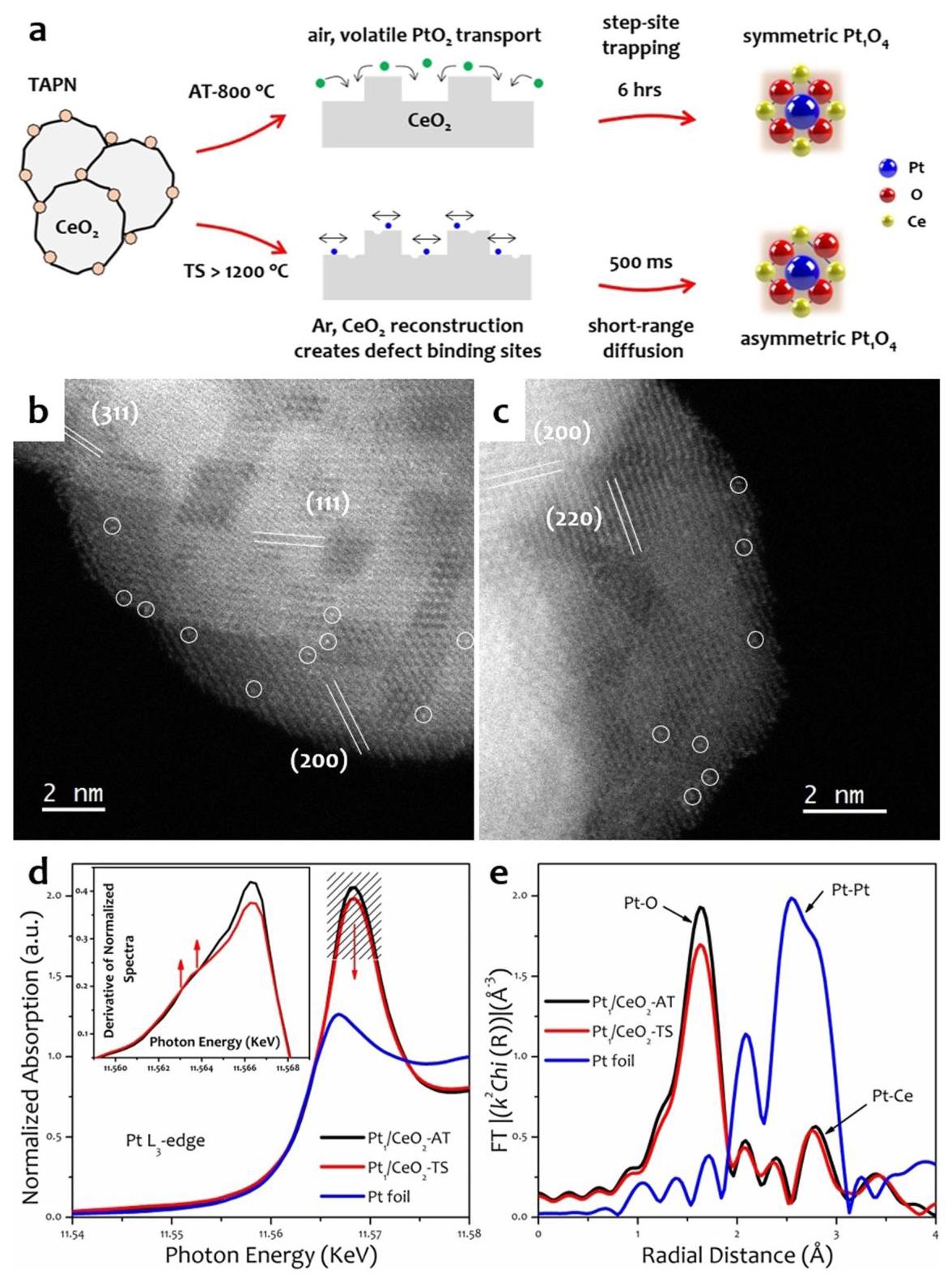
分析结果: 在AT和TS催化剂中均只观察到孤立的Pt原子,没有发现Pt团簇或纳米颗粒,证明了两种方法均成功实现了Pt的单原子分散。
图1d和1e的XAS谱图提供了Pt电子结构和局部配位的信息。
分析结果(XANES,图1d): TS催化剂的白线强度略低于AT催化剂,表明其Pt价态稍低或配位对称性降低。TS谱图在吸收边附近出现了一个上升边特征(导数曲线中的凸起),这通常与Pt位点局部配位对称性的降低有关,证实了不对称Pt1O4配位的存在。
分析结果(EXAFS,图1e和表1): AT催化剂的EXAFS数据可以用四个等效的Pt-O键(~1.995 Å)完美拟合,符合对称的Pt1O4配位。TS催化剂的EXAFS数据则需用三个较短的Pt-OS键(~1.979 Å)和一个较长的Pt-OL键(~2.051 Å)来拟合,证实了不对称Pt1O4配位结构的形成。OL可能来源于表面羟基或化学吸附的氧物种。
图表分析:催化性能与反应机理
图2a比较了AT、TS催化剂和空白CeO2的CO氧化起燃曲线。
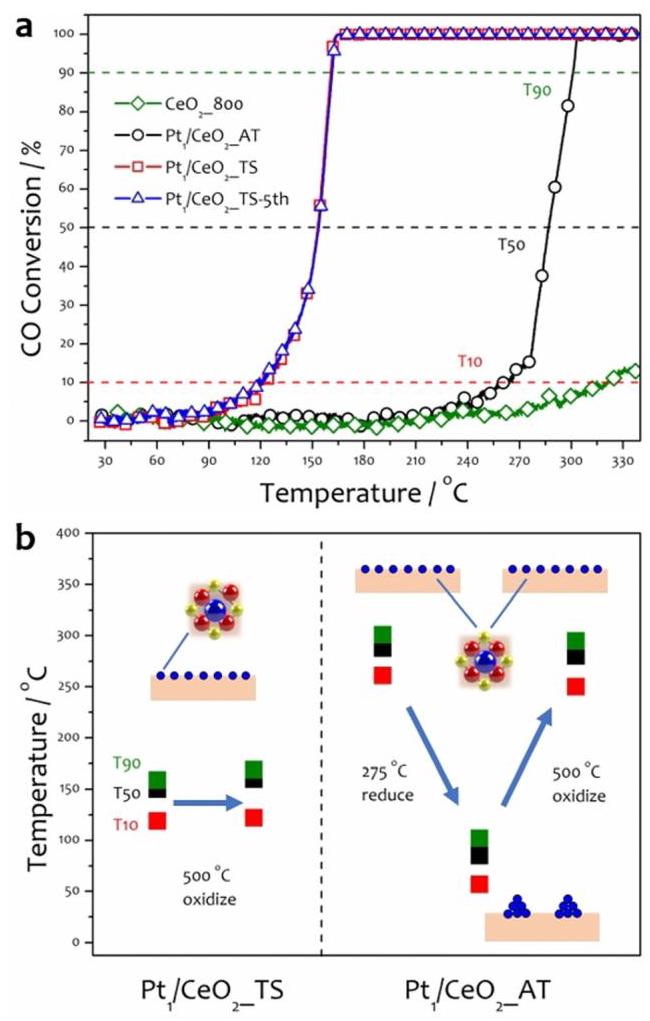
分析结果: AT催化剂在200°C以下几乎没有活性,其活性在240°C以上才优于空白CeO2。相比之下,TS催化剂表现出优异的低温活性,其T50(~150°C)比AT催化剂(~287°C)低了约137°C,并且经过多次循环测试后活性保持稳定。
图2b展示了催化剂在经过还原和氧化处理后的T10, T50, T90变化。
分析结果: AT催化剂经还原处理后(形成Pt NPs),T10, T50, T90显著降低(活性提高),但随后在500°C氧化处理后,这些温度值几乎回到原始水平(活性急剧下降)。TS催化剂在500°C氧化处理后,T10, T50, T90仅略有升高(2-10°C),表现出卓越的抗氧化失活稳定性。
图3展示了原位DRIFTS实验结果,用于探测反应条件下Pt物种的演变。
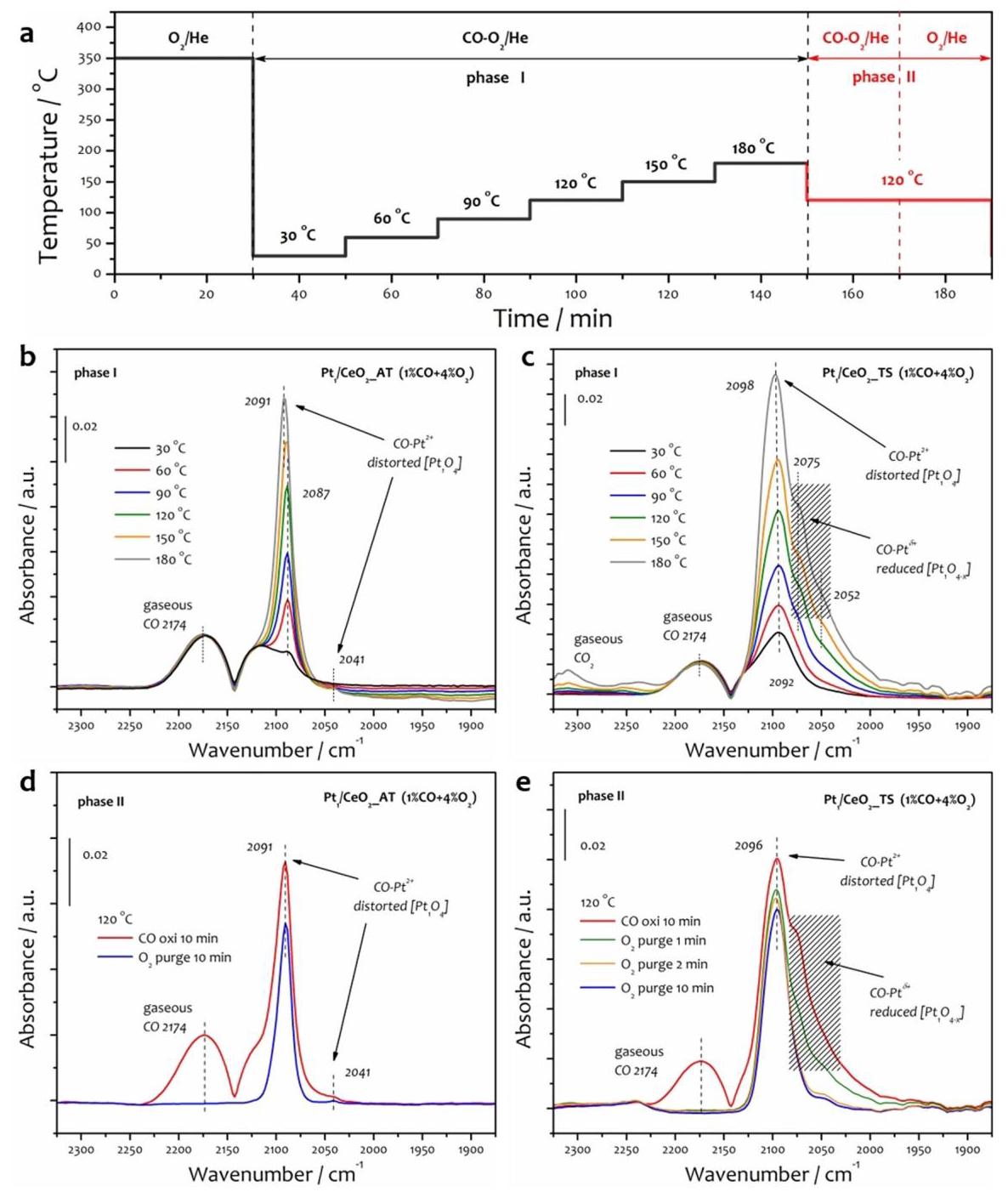
分析结果(Phase-I, 图3b,c):
- AT催化剂(图3b):在30°C仅观察到非常弱的~2087 cm-1峰(归属于线性吸附在Pt2+上的CO),表明对称Pt1O4难以吸附CO。随温度升高,该峰强度增加且蓝移,表明配位结构发生扭曲。
- TS催化剂(图3c):在30°C就观察到更强、更对称的~2092 cm-1峰,证实了不对称Pt1O4结构的存在(CO吸附更强)。在温度≥90°C时,出现了~2075 cm-1和~2052 cm-1的两个肩峰,这些峰不属于金属Pt0或氧化PtOx团簇,而被指认为部分还原的Pt1δ+物种(Pt1O4-x配位)上吸附的CO。
分析结果(Phase-II, 图3d,e): 在120°C切换为O2/He吹扫后,AT催化剂上的CO-Pt2+峰仅轻微减弱(图3d),与其低温活性差相符。TS催化剂上的CO-Pt2+峰缓慢减弱,而CO-Pt1δ+肩峰(2075, 2052 cm-1)则在2分钟内迅速消失(图3e),表明部分还原的Pt1δ+物种具有极高的反应活性,是低温CO氧化的活性中心。
图4总结了TS催化剂中Pt单原子在反应过程中的动态演变示意图。
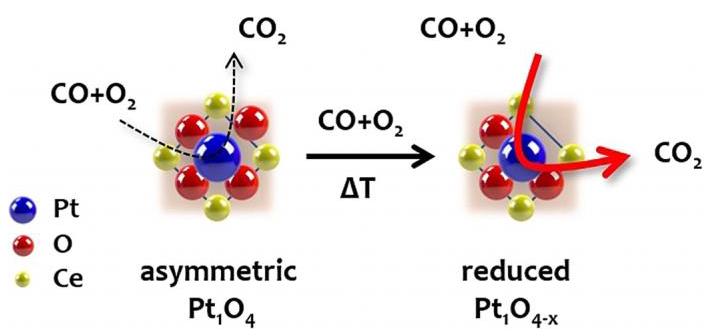
分析结果: TS合成创造了不对称的Pt1O4配位起点。在CO氧化反应条件下,Pt2+动态演变为部分还原的Pt1δ+物种(Pt1O4-x配位),这些物种极大地促进了低温CO氧化。重要的是,这个过程是可逆的,避免了形成易失活的金属Pt NPs,从而实现了高活性和高稳定性的统一。